Manipulation et analyse de données avec dplyr
Dernière mise à jour le 2024-09-12 | Modifier cette page
Vue d'ensemble
Questions
- Analyse de données dans R à l’aide du méta-paquet Tidyverse
Objectifs
- Décrivez l’objectif des packages
dplyrettidyr. - Décrivez plusieurs de leurs fonctions extrêmement utiles pour manipuler des données.
- Décrivez le concept d’un format de tableau large et long, et voyez comment remodeler un bloc de données d’un format à l’autre.
- Montrez comment joindre des tables.
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Manipulation des données à l’aide de
dplyr et
tidyr
Le sous-ensemble entre crochets est pratique, mais il peut être fastidieux et difficile à lire, en particulier pour les opérations compliquées.
Certains packages peuvent grandement faciliter notre tâche lorsque
nous manipulons des données. Les packages dans R sont essentiellement
des ensembles de fonctions supplémentaires qui vous permettent de faire
plus de choses. Les fonctions que nous avons utilisées jusqu’à présent,
comme str() ou data.frame(), sont intégrées à
R ; Le chargement de packages peut vous donner accès à d’autres
fonctions spécifiques. Avant d’utiliser un package pour la première
fois, vous devez l’installer sur votre machine, puis vous devez
l’importer à chaque session R suivante lorsque vous en avez besoin.
Le package
dplyrfournit des outils puissants pour les tâches de manipulation de données. Il est conçu pour fonctionner directement avec des trames de données, avec de nombreuses tâches de manipulation optimisées.Comme nous le verrons plus loin, nous souhaitons parfois qu’un bloc de données soit remodelé pour pouvoir effectuer des analyses spécifiques ou pour la visualisation. Le package
tidyrrésout ce problème courant de remodelage des données et fournit des outils pour manipuler les données de manière ordonnée.
Pour en savoir plus sur dplyr et
tidyr après l’atelier, vous voudrez
peut-être consulter ceci transformation de données pratique avec ** et
ceci celui sur .
- Le package
tidyverseest un “package parapluie” qui installe plusieurs packages utiles pour l’analyse des données qui fonctionnent bien ensemble, tels quetidyr, * *dplyr**,ggplot2,tibble, etc. Ces packages nous aident à travailler et à interagir avec les données. Ils nous permettent de faire beaucoup de choses avec vos données, comme le sous-ensemble, la transformation, la visualisation, etc.
Si vous avez effectué la configuration, vous devriez déjà avoir installé le package Tidyverse. Vérifiez si vous l’avez en essayant de le charger depuis la bibliothèque :
R
## load the tidyverse packages, incl. dplyr
library("tidyverse")
Si vous recevez un message d’erreur
il n'y a pas de package appelé 'tidyverse' alors vous
n’avez pas installé le package pour cette version de R. Pour installer
le package tidyverse, tapez :
R
BiocManager::install("tidyverse")
Si vous avez dû installer le package
tidyverse, n’oubliez pas de le charger
dans cette session R en utilisant la commande library()
ci-dessus !
Chargement de données avec Tidyverse
Instead of read.csv(), we will read in our data using
the read_csv() function (notice the _ instead
of the .), from the tidyverse package
readr.
R
rna <- read_csv("data/rnaseq.csv")
## view the data
rna
SORTIE
# A tibble: 32,428 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Notez que la classe des données est désormais appelée « tibble ».
Tibbles modifie certains des comportements des objets de trame de données que nous avons introduits dans le précédemment. La structure des données est très similaire à une trame de données. Pour nos besoins , les seules différences sont les suivantes :
Il affiche le type de données de chaque colonne sous son nom. Notez que <
dbl> est un type de données défini pour contenir des valeurs numériques avec points décimaux.Il imprime uniquement les premières lignes de données et seulement autant de colonnes que peuvent contenir un écran.
Nous allons maintenant apprendre certaines des fonctions
dplyr les plus courantes :
-
select(): sous-ensemble de colonnes -
filter(): sous-ensemble de lignes sur conditions -
mutate(): crée de nouvelles colonnes en utilisant les informations d’autres colonnes -
group_by()etsummarise(): créent des statistiques récapitulatives sur des données groupées -
arrange(): trier les résultats -
count(): compte les valeurs discrètes
Sélection de colonnes et filtrage de lignes
Pour sélectionner les colonnes d’un bloc de données, utilisez
select(). Le premier argument de cette fonction est la
trame de données (rna), et les arguments suivants sont les
colonnes à conserver.
R
select(rna, gene, sample, tissue, expression)
SORTIE
# A tibble: 32,428 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545336 Cerebellum 1170
2 Apod GSM2545336 Cerebellum 36194
3 Cyp2d22 GSM2545336 Cerebellum 4060
4 Klk6 GSM2545336 Cerebellum 287
5 Fcrls GSM2545336 Cerebellum 85
6 Slc2a4 GSM2545336 Cerebellum 782
7 Exd2 GSM2545336 Cerebellum 1619
8 Gjc2 GSM2545336 Cerebellum 288
9 Plp1 GSM2545336 Cerebellum 43217
10 Gnb4 GSM2545336 Cerebellum 1071
# ℹ 32,418 more rowsPour sélectionner toutes les colonnes sauf certaines, mettez un “-” devant la variable pour l’exclure.
R
select(rna, -tissue, -organism)
SORTIE
# A tibble: 32,428 × 17
gene sample expression age sex infection strain time mouse ENTREZID
<chr> <chr> <dbl> <dbl> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Asl GSM2545… 1170 8 Fema… Influenz… C57BL… 8 14 109900
2 Apod GSM2545… 36194 8 Fema… Influenz… C57BL… 8 14 11815
3 Cyp2d22 GSM2545… 4060 8 Fema… Influenz… C57BL… 8 14 56448
4 Klk6 GSM2545… 287 8 Fema… Influenz… C57BL… 8 14 19144
5 Fcrls GSM2545… 85 8 Fema… Influenz… C57BL… 8 14 80891
6 Slc2a4 GSM2545… 782 8 Fema… Influenz… C57BL… 8 14 20528
7 Exd2 GSM2545… 1619 8 Fema… Influenz… C57BL… 8 14 97827
8 Gjc2 GSM2545… 288 8 Fema… Influenz… C57BL… 8 14 118454
9 Plp1 GSM2545… 43217 8 Fema… Influenz… C57BL… 8 14 18823
10 Gnb4 GSM2545… 1071 8 Fema… Influenz… C57BL… 8 14 14696
# ℹ 32,418 more rows
# ℹ 7 more variables: product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>Cela sélectionnera toutes les variables de rna sauf
tissu et organism.
Pour choisir des lignes en fonction d’un critère spécifique, utilisez
filter() :
R
filter(rna, sex == "Male")
SORTIE
# A tibble: 14,740 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 626 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
2 Apod GSM254… 13021 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
3 Cyp2d22 GSM254… 2171 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
4 Klk6 GSM254… 448 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
5 Fcrls GSM254… 180 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
6 Slc2a4 GSM254… 313 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
7 Exd2 GSM254… 2366 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
8 Gjc2 GSM254… 310 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
9 Plp1 GSM254… 53126 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
10 Gnb4 GSM254… 1355 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
# ℹ 14,730 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>R
filter(rna, sex == "Male" & infection == "NonInfected")
SORTIE
# A tibble: 4,422 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 535 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
2 Apod GSM254… 13668 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
3 Cyp2d22 GSM254… 2008 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
4 Klk6 GSM254… 1101 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
5 Fcrls GSM254… 375 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
6 Slc2a4 GSM254… 249 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
7 Exd2 GSM254… 3126 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
8 Gjc2 GSM254… 791 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
9 Plp1 GSM254… 98658 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
10 Gnb4 GSM254… 2437 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
# ℹ 4,412 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Imaginons maintenant que nous nous intéressions aux homologues
humains des gènes de souris analysés dans cet ensemble de données. Ces
informations se trouvent dans la dernière colonne du tibble
rna, nommée
hsapiens_homolog_associated_gene_name. Pour le visualiser
facilement, nous allons créer un nouveau tableau contenant uniquement
les 2 colonnes gene et
hsapiens_homolog_associated_gene_name.
R
genes <- select(rna, gene, hsapiens_homolog_associated_gene_name)
genes
SORTIE
# A tibble: 32,428 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Asl ASL
2 Apod APOD
3 Cyp2d22 CYP2D6
4 Klk6 KLK6
5 Fcrls FCRL2
6 Slc2a4 SLC2A4
7 Exd2 EXD2
8 Gjc2 GJC2
9 Plp1 PLP1
10 Gnb4 GNB4
# ℹ 32,418 more rowsCertains gènes de souris n’ont pas d’homologues humains. Ceux-ci
peuvent être récupérés en utilisant filter() et la fonction
is.na(), qui détermine si quelque chose est un
NA.
R
filter(genes, is.na(hsapiens_homolog_associated_gene_name))
SORTIE
# A tibble: 4,290 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Prodh <NA>
2 Tssk5 <NA>
3 Vmn2r1 <NA>
4 Gm10654 <NA>
5 Hexa <NA>
6 Sult1a1 <NA>
7 Gm6277 <NA>
8 Tmem198b <NA>
9 Adam1a <NA>
10 Ebp <NA>
# ℹ 4,280 more rowsSi on veut conserver uniquement les gènes de souris qui ont un
homologue humain, on peut insérer un “!” symbole qui annule le résultat,
nous demandons donc chaque ligne où
hsapiens_homolog_associated_gene_name n’est pas un
NA.
R
filter(genes, !is.na(hsapiens_homolog_associated_gene_name))
SORTIE
# A tibble: 28,138 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Asl ASL
2 Apod APOD
3 Cyp2d22 CYP2D6
4 Klk6 KLK6
5 Fcrls FCRL2
6 Slc2a4 SLC2A4
7 Exd2 EXD2
8 Gjc2 GJC2
9 Plp1 PLP1
10 Gnb4 GNB4
# ℹ 28,128 more rowsTuyaux
Et si vous souhaitez sélectionner et filtrer en même temps ? Il existe trois façons de procéder : utiliser des étapes intermédiaires, des fonctions imbriquées ou des tuyaux.
Avec des étapes intermédiaires, vous créez un bloc de données temporaire et l’utilisez comme entrée de la fonction suivante, comme ceci :
R
rna2 <- filter(rna, sex == "Male")
rna3 <- select(rna2, gene, sample, tissue, expression)
rna3
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsCeci est lisible, mais peut encombrer votre espace de travail avec de nombreux objets intermédiaires que vous devez nommer individuellement. Avec plusieurs étapes , cela peut être difficile à suivre.
Vous pouvez également imbriquer des fonctions (c’est-à-dire une fonction dans une autre), comme ceci :
R
rna3 <- select(filter(rna, sex == "Male"), gene, sample, tissue, expression)
rna3
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsC’est pratique, mais peut être difficile à lire si trop de fonctions sont imbriquées, car R évalue l’expression de l’intérieur vers l’extérieur (dans ce cas, filtrer, puis sélectionner).
La dernière option, pipes, est un ajout récent à R. Pipes vous permet de prendre la sortie d’une fonction et de l’envoyer directement à la suivante, ce qui est utile lorsque vous devez faire beaucoup de choses dans le même ensemble de données.
Les tuyaux dans R ressemblent à %>% (mis à
disposition via le package magrittr ) ou
|> (via la base R). If you use RStudio, you can type the
pipe with Ctrl + Shift + M if you have
a PC or Cmd + Shift + M if you have a
Mac.
Dans le code ci-dessus, nous utilisons le tube pour envoyer
l’ensemble de données rna d’abord via filter()
pour conserver les lignes où sex est Homme, puis via
select() pour conserver uniquement les colonnes
gène, échantillon, tissu et
expression.
Le tube %>% prend l’objet à sa gauche et le passe
directement comme le premier argument de la fonction à sa droite, nous
n’avons pas besoin de inclure explicitement le bloc de données comme un
argument pour les fonctions filter() et
select().
R
rna %>%
filter(sex == "Male") %>%
select(gene, sample, tissue, expression)
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsCertains trouveront peut-être utile de lire le tube comme le mot «
alors ». Par exemple, dans l’exemple ci-dessus, nous avons pris la trame
de données rna, puis nous avons
filtré pour les lignes avec sex == "Male",
puis nous avons « sélectionné les colonnes « gène », «
échantillon », « tissu » et « expression ».
Les fonctions dplyr en elles-mêmes sont
quelque peu simples, mais en les combinant dans des flux de travail
linéaires avec le tube, nous pouvons accomplir des manipulations plus
complexes de trames de données.
Si nous voulons créer un nouvel objet avec cette version plus petite des données, nous pouvons lui attribuer un nouveau nom :
R
rna3 <- rna %>%
filter(sex == "Male") %>%
select(gene, sample, tissue, expression)
rna3
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsR
rna %>%
filter(expression > 50000,
sex == "Female",
time == 0 ) %>%
select(gene, sample, time, expression, age)
SORTIE
# A tibble: 9 × 5
gene sample time expression age
<chr> <chr> <dbl> <dbl> <dbl>
1 Plp1 GSM2545337 0 101241 8
2 Atp1b1 GSM2545337 0 53260 8
3 Plp1 GSM2545338 0 96534 8
4 Atp1b1 GSM2545338 0 50614 8
5 Plp1 GSM2545348 0 102790 8
6 Atp1b1 GSM2545348 0 59544 8
7 Plp1 GSM2545353 0 71237 8
8 Glul GSM2545353 0 52451 8
9 Atp1b1 GSM2545353 0 61451 8Subir une mutation
Vous souhaiterez fréquemment créer de nouvelles colonnes basées sur
les valeurs des colonnes existantes, par exemple pour effectuer des
conversions d’unités ou pour trouver le rapport des valeurs dans deux
colonnes . Pour cela, nous utiliserons mutate().
Pour créer une nouvelle colonne de temps en heures :
R
rna %>%
mutate(time_hours = time * 24) %>%
select(time, time_hours)
SORTIE
# A tibble: 32,428 × 2
time time_hours
<dbl> <dbl>
1 8 192
2 8 192
3 8 192
4 8 192
5 8 192
6 8 192
7 8 192
8 8 192
9 8 192
10 8 192
# ℹ 32,418 more rowsVous pouvez également créer une deuxième nouvelle colonne basée sur
la première nouvelle colonne dans le même appel de
mutate() :
R
rna %>%
mutate(time_hours = time * 24,
time_mn = time_hours * 60) %>%
select(time, time_hours, time_mn)
SORTIE
# A tibble: 32,428 × 3
time time_hours time_mn
<dbl> <dbl> <dbl>
1 8 192 11520
2 8 192 11520
3 8 192 11520
4 8 192 11520
5 8 192 11520
6 8 192 11520
7 8 192 11520
8 8 192 11520
9 8 192 11520
10 8 192 11520
# ℹ 32,418 more rowsDéfi
Créez un nouveau bloc de données à partir des données
rna qui répond aux critères suivants : contient uniquement
le gène, le nom_chromosome,
phenotype_description, sample et
expression Colonnes. Les valeurs de l’expression doivent
être transformées en log. Cette trame de données doit contenir
uniquement des gènes situés sur les chromosomes sexuels, associés à un
phénotype _description, et avec une expression log supérieure à 5.
Astuce : réfléchissez à la façon dont les commandes doivent être ordonnées pour produire ce bloc de données !
R
rna %>%
mutate(expression = log(expression)) %>%
select(gene, chromosome_name, phenotype_description, sample, expression) %>%
filter(chromosome_name == "X" | chromosome_name == "Y") %>%
filter(!is.na(phenotype_description)) %>%
filter(expression > 5)
SORTIE
# A tibble: 649 × 5
gene chromosome_name phenotype_description sample expression
<chr> <chr> <chr> <chr> <dbl>
1 Plp1 X abnormal CNS glial cell morphology GSM25… 10.7
2 Slc7a3 X decreased body length GSM25… 5.46
3 Plxnb3 X abnormal coat appearance GSM25… 6.58
4 Rbm3 X abnormal liver morphology GSM25… 9.32
5 Cfp X abnormal cardiovascular system phys… GSM25… 6.18
6 Ebp X abnormal embryonic erythrocyte morp… GSM25… 6.68
7 Cd99l2 X abnormal cellular extravasation GSM25… 8.04
8 Piga X abnormal brain development GSM25… 6.06
9 Pim2 X decreased T cell proliferation GSM25… 7.11
10 Itm2a X no abnormal phenotype detected GSM25… 7.48
# ℹ 639 more rowsAnalyse de données fractionnée-appliquée-combinée
De nombreuses tâches d’analyse de données peuvent être abordées à
l’aide du paradigme split-apply-combine : divisez les données
en groupes, appliquez une analyse à chaque groupe, puis combinez les
résultats. dplyr rend cela très facile
grâce à l’utilisation de la fonction group_by().
R
rna %>%
group_by(gene)
SORTIE
# A tibble: 32,428 × 19
# Groups: gene [1,474]
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>La fonction group_by() n’effectue aucun traitement de
données, elle regroupe les données en sous-ensembles : dans l’exemple
ci-dessus, notre tibble initial de 32428 les observations
sont divisées en 1474 en fonction de la variable gene.
On pourrait de même décider de regrouper les tibbles par échantillons :
R
rna %>%
group_by(sample)
SORTIE
# A tibble: 32,428 × 19
# Groups: sample [22]
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Ici, notre tibble initial d’observations 32428 est
divisé en groupes 22 en fonction de la variable sample.
Une fois les données regroupées, les opérations suivantes seront appliquées sur chaque groupe indépendamment.
La fonction summaris()
group_by() est souvent utilisé avec
summarise(), qui réduit chaque groupe en un résumé sur une
seule ligne de ce groupe.
group_by() prend comme arguments les noms de colonnes
qui contiennent les variables catégorielles pour
lesquelles vous souhaitez calculer les statistiques récapitulatives .
Donc, pour calculer l’expression moyenne par gène :
R
rna %>%
group_by(gene) %>%
summarise(mean_expression = mean(expression))
SORTIE
# A tibble: 1,474 × 2
gene mean_expression
<chr> <dbl>
1 AI504432 1053.
2 AW046200 131.
3 AW551984 295.
4 Aamp 4751.
5 Abca12 4.55
6 Abcc8 2498.
7 Abhd14a 525.
8 Abi2 4909.
9 Abi3bp 1002.
10 Abl2 2124.
# ℹ 1,464 more rowsNous pourrions également vouloir calculer les niveaux d’expression moyens de tous les gènes dans chaque échantillon :
R
rna %>%
group_by(sample) %>%
summarise(mean_expression = mean(expression))
SORTIE
# A tibble: 22 × 2
sample mean_expression
<chr> <dbl>
1 GSM2545336 2062.
2 GSM2545337 1766.
3 GSM2545338 1668.
4 GSM2545339 1696.
5 GSM2545340 1682.
6 GSM2545341 1638.
7 GSM2545342 1594.
8 GSM2545343 2107.
9 GSM2545344 1712.
10 GSM2545345 1700.
# ℹ 12 more rowsMais on peut aussi regrouper par plusieurs colonnes :
R
rna %>%
group_by(gene, infection, time) %>%
summarise(mean_expression = mean(expression))
SORTIE
`summarise()` has grouped output by 'gene', 'infection'. You can override using
the `.groups` argument.SORTIE
# A tibble: 4,422 × 4
# Groups: gene, infection [2,948]
gene infection time mean_expression
<chr> <chr> <dbl> <dbl>
1 AI504432 InfluenzaA 4 1104.
2 AI504432 InfluenzaA 8 1014
3 AI504432 NonInfected 0 1034.
4 AW046200 InfluenzaA 4 152.
5 AW046200 InfluenzaA 8 81
6 AW046200 NonInfected 0 155.
7 AW551984 InfluenzaA 4 302.
8 AW551984 InfluenzaA 8 342.
9 AW551984 NonInfected 0 238
10 Aamp InfluenzaA 4 4870
# ℹ 4,412 more rowsUne fois les données regroupées, vous pouvez également résumer plusieurs variables en même temps (et pas nécessairement sur la même variable). Par exemple, nous pourrions ajouter une colonne indiquant l’expression médiane par gène et par condition :
R
rna %>%
group_by(gene, infection, time) %>%
summarise(mean_expression = mean(expression),
median_expression = median(expression))
SORTIE
`summarise()` has grouped output by 'gene', 'infection'. You can override using
the `.groups` argument.SORTIE
# A tibble: 4,422 × 5
# Groups: gene, infection [2,948]
gene infection time mean_expression median_expression
<chr> <chr> <dbl> <dbl> <dbl>
1 AI504432 InfluenzaA 4 1104. 1094.
2 AI504432 InfluenzaA 8 1014 985
3 AI504432 NonInfected 0 1034. 1016
4 AW046200 InfluenzaA 4 152. 144.
5 AW046200 InfluenzaA 8 81 82
6 AW046200 NonInfected 0 155. 163
7 AW551984 InfluenzaA 4 302. 245
8 AW551984 InfluenzaA 8 342. 287
9 AW551984 NonInfected 0 238 265
10 Aamp InfluenzaA 4 4870 4708
# ℹ 4,412 more rowsR
rna %>%
filter(gene == "Dok3") %>%
group_by(time) %>%
summarise(mean = mean(expression))
SORTIE
# A tibble: 3 × 2
time mean
<dbl> <dbl>
1 0 169
2 4 156.
3 8 61 Compte
Lorsque nous travaillons avec des données, nous souhaitons souvent
connaître le nombre d’observations trouvées pour chaque facteur ou
combinaison de facteurs. Pour cette tâche,
dplyr fournit count(). Par
exemple, si nous voulions compter le nombre de lignes de données pour
chaque échantillon infecté et non infecté, nous ferions :
R
rna %>%
count(infection)
SORTIE
# A tibble: 2 × 2
infection n
<chr> <int>
1 InfluenzaA 22110
2 NonInfected 10318La fonction count() est un raccourci pour quelque chose
que nous avons déjà vu : regrouper par une variable et le résumer en
comptant le nombre d’observations dans ce groupe. En d’autres termes,
« rna %>% count(infection) » équivaut à :
R
rna %>%
group_by(infection) %>%
summarise(n = n())
SORTIE
# A tibble: 2 × 2
infection n
<chr> <int>
1 InfluenzaA 22110
2 NonInfected 10318L’exemple précédent montre l’utilisation de count() pour
compter le nombre de lignes/observations pour un facteur
(c’est-à-dire infection). Si nous voulions compter une
combinaison de facteurs, telle que infection et
time, nous spécifierions le premier et le deuxième facteur
comme arguments de count() :
R
rna %>%
count(infection, time)
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318ce qui équivaut à ceci :
R
rna %>%
group_by(infection, time) %>%
summarise(n = n())
SORTIE
`summarise()` has grouped output by 'infection'. You can override using the
`.groups` argument.SORTIE
# A tibble: 3 × 3
# Groups: infection [2]
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318Il est parfois utile de trier le résultat pour faciliter les
comparaisons. Nous pouvons utiliser arrange() pour trier le
tableau. Par exemple, nous pourrions vouloir organiser le tableau
ci-dessus par heure :
R
rna %>%
count(infection, time) %>%
arrange(time)
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 NonInfected 0 10318
2 InfluenzaA 4 11792
3 InfluenzaA 8 10318ou par comptages :
R
rna %>%
count(infection, time) %>%
arrange(n)
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 8 10318
2 NonInfected 0 10318
3 InfluenzaA 4 11792Pour trier par ordre décroissant, nous devons ajouter la fonction
desc() :
R
rna %>%
count(infection, time) %>%
arrange(desc(n))
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318Défi
- Combien de gènes ont été analysés dans chaque échantillon ?
- Utilisez
group_by()etsummarise()pour évaluer la profondeur de séquençage (la somme de tous les comptes) dans chaque échantillon. Quel échantillon a la profondeur de séquençage la plus élevée ? - Choisissez un échantillon et évaluez le nombre de gènes par biotype.
- Identifiez les gènes associés à la description du phénotype « méthylation anormale de l’ADN » et calculez leur expression moyenne (en log) au temps 0, au temps 4 et au temps 8.
R
## 1.
rna %>%
count(sample)
SORTIE
# A tibble: 22 × 2
sample n
<chr> <int>
1 GSM2545336 1474
2 GSM2545337 1474
3 GSM2545338 1474
4 GSM2545339 1474
5 GSM2545340 1474
6 GSM2545341 1474
7 GSM2545342 1474
8 GSM2545343 1474
9 GSM2545344 1474
10 GSM2545345 1474
# ℹ 12 more rowsR
## 2.
rna %>%
group_by(sample) %>%
summarise(seq_depth = sum(expression)) %>%
arrange(desc(seq_depth))
SORTIE
# A tibble: 22 × 2
sample seq_depth
<chr> <dbl>
1 GSM2545350 3255566
2 GSM2545352 3216163
3 GSM2545343 3105652
4 GSM2545336 3039671
5 GSM2545380 3036098
6 GSM2545353 2953249
7 GSM2545348 2913678
8 GSM2545362 2913517
9 GSM2545351 2782464
10 GSM2545349 2758006
# ℹ 12 more rowsR
## 3.
rna %>%
filter(sample == "GSM2545336") %>%
count(gene_biotype) %>%
arrange(desc(n))
SORTIE
# A tibble: 13 × 2
gene_biotype n
<chr> <int>
1 protein_coding 1321
2 lncRNA 69
3 processed_pseudogene 59
4 miRNA 7
5 snoRNA 5
6 TEC 4
7 polymorphic_pseudogene 2
8 unprocessed_pseudogene 2
9 IG_C_gene 1
10 scaRNA 1
11 transcribed_processed_pseudogene 1
12 transcribed_unitary_pseudogene 1
13 transcribed_unprocessed_pseudogene 1R
## 4.
rna %>%
filter(phenotype_description == "abnormal DNA methylation") %>%
group_by(gene, time) %>%
summarise(mean_expression = mean(log(expression))) %>%
arrange()
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 6 × 3
# Groups: gene [2]
gene time mean_expression
<chr> <dbl> <dbl>
1 Xist 0 6.95
2 Xist 4 6.34
3 Xist 8 7.13
4 Zdbf2 0 6.27
5 Zdbf2 4 6.27
6 Zdbf2 8 6.19Remodeler les données
Dans le tibble rna, les lignes contiennent des valeurs
d’expression (l’unité) qui sont associées à une combinaison de 2 autres
variables : gene et sample.
Toutes les autres colonnes correspondent à des variables décrivant soit l’échantillon (organisme, âge, sexe, …) soit le gène (gène_biotype, ENTREZ_ID, produit, …). Les variables qui ne changent pas avec les gènes ou avec les échantillons auront la même valeur dans toutes les lignes.
R
rna %>%
arrange(gene)
SORTIE
# A tibble: 32,428 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 AI504432 GSM25… 1230 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 AI504432 GSM25… 1085 Mus mus… 8 Fema… NonInfec… C57BL… 0 Cereb…
3 AI504432 GSM25… 969 Mus mus… 8 Fema… NonInfec… C57BL… 0 Cereb…
4 AI504432 GSM25… 1284 Mus mus… 8 Fema… Influenz… C57BL… 4 Cereb…
5 AI504432 GSM25… 966 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
6 AI504432 GSM25… 918 Mus mus… 8 Male Influenz… C57BL… 8 Cereb…
7 AI504432 GSM25… 985 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 AI504432 GSM25… 972 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
9 AI504432 GSM25… 1000 Mus mus… 8 Fema… Influenz… C57BL… 4 Cereb…
10 AI504432 GSM25… 816 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Cette structure est appelée « format long », car une colonne contient toutes les valeurs, et d’autres colonnes répertorient le contexte de la valeur.
Dans certains cas, le « format long » n’est pas vraiment « lisible par l’homme », et un autre format, un « format large » est préféré, comme manière plus compacte de représenter les données. This is typically the case with gene expression values that scientists are used to look as matrices, were rows represent genes and columns represent samples.
Dans ce format, il deviendrait donc simple d’explorer la relation entre les niveaux d’expression génique au sein et entre les échantillons.
SORTIE
# A tibble: 1,474 × 23
gene GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988
2 Apod 36194 10347 9173 10620 13021 29594
3 Cyp2d22 4060 1616 1603 1901 2171 3349
4 Klk6 287 629 641 578 448 195
5 Fcrls 85 233 244 237 180 38
6 Slc2a4 782 231 248 265 313 786
7 Exd2 1619 2288 2235 2513 2366 1359
8 Gjc2 288 595 568 551 310 146
9 Plp1 43217 101241 96534 58354 53126 27173
10 Gnb4 1071 1791 1867 1430 1355 798
# ℹ 1,464 more rows
# ℹ 16 more variables: GSM2545342 <dbl>, GSM2545343 <dbl>, GSM2545344 <dbl>,
# GSM2545345 <dbl>, GSM2545346 <dbl>, GSM2545347 <dbl>, GSM2545348 <dbl>,
# GSM2545349 <dbl>, GSM2545350 <dbl>, GSM2545351 <dbl>, GSM2545352 <dbl>,
# GSM2545353 <dbl>, GSM2545354 <dbl>, GSM2545362 <dbl>, GSM2545363 <dbl>,
# GSM2545380 <dbl>Pour convertir les valeurs d’expression génique de rna
en un format large, nous devons créer une nouvelle table où les valeurs
de la colonne sample deviendraient les noms des variables
de colonne.
The key point here is that we are still following a tidy data structure, but we have reshaped the data according to the observations of interest: expression levels per gene instead of recording them per gene and per sample.
La transformation inverse serait de transformer les noms de colonnes en valeurs d’une nouvelle variable.
Nous pouvons effectuer ces deux transformations avec deux fonctions
tidyr, pivot_longer() et
pivot_wider() (voir ici
pour détails).
Pivoter les données dans un format plus large
Sélectionnons les 3 premières colonnes de rna et
utilisons pivot_wider() pour transformer les données en
grand format.
R
rna_exp <- rna %>%
select(gene, sample, expression)
rna_exp
SORTIE
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Cyp2d22 GSM2545336 4060
4 Klk6 GSM2545336 287
5 Fcrls GSM2545336 85
6 Slc2a4 GSM2545336 782
7 Exd2 GSM2545336 1619
8 Gjc2 GSM2545336 288
9 Plp1 GSM2545336 43217
10 Gnb4 GSM2545336 1071
# ℹ 32,418 more rowspivot_wider prend trois arguments principaux :
- les données à transformer ;
- le
names_from: la colonne dont les valeurs deviendront de nouveaux noms de colonne ; - les
values_from: la colonne dont les valeurs rempliront les nouvelles colonnes .
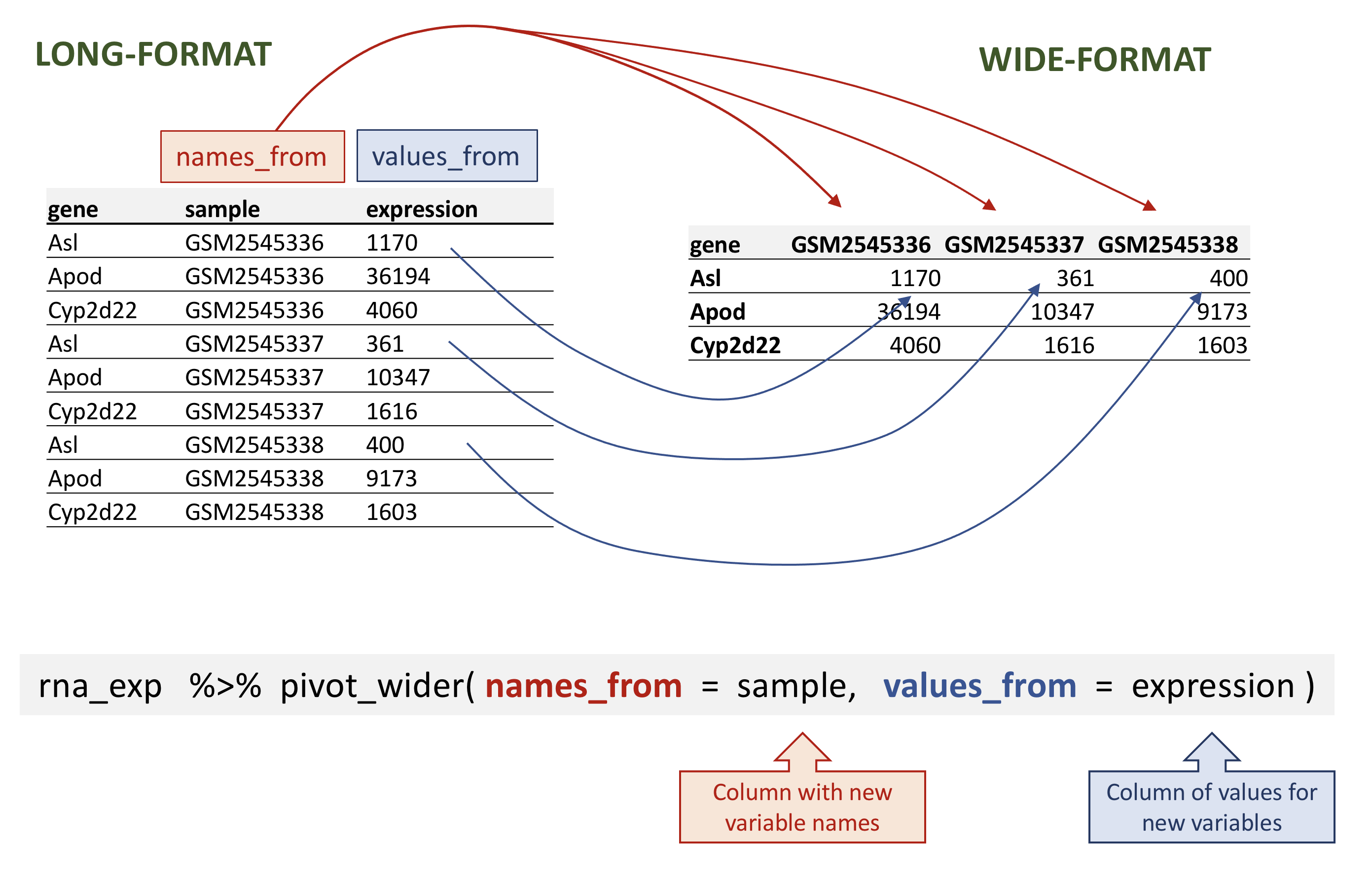
rna.
R
rna_wide <- rna_exp %>%
pivot_wider(names_from = sample,
values_from = expression)
rna_wide
SORTIE
# A tibble: 1,474 × 23
gene GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988
2 Apod 36194 10347 9173 10620 13021 29594
3 Cyp2d22 4060 1616 1603 1901 2171 3349
4 Klk6 287 629 641 578 448 195
5 Fcrls 85 233 244 237 180 38
6 Slc2a4 782 231 248 265 313 786
7 Exd2 1619 2288 2235 2513 2366 1359
8 Gjc2 288 595 568 551 310 146
9 Plp1 43217 101241 96534 58354 53126 27173
10 Gnb4 1071 1791 1867 1430 1355 798
# ℹ 1,464 more rows
# ℹ 16 more variables: GSM2545342 <dbl>, GSM2545343 <dbl>, GSM2545344 <dbl>,
# GSM2545345 <dbl>, GSM2545346 <dbl>, GSM2545347 <dbl>, GSM2545348 <dbl>,
# GSM2545349 <dbl>, GSM2545350 <dbl>, GSM2545351 <dbl>, GSM2545352 <dbl>,
# GSM2545353 <dbl>, GSM2545354 <dbl>, GSM2545362 <dbl>, GSM2545363 <dbl>,
# GSM2545380 <dbl>Notez que par défaut, la fonction pivot_wider() ajoutera
NA pour les valeurs manquantes.
Imaginons que, pour une raison quelconque, nous ayons des valeurs d’expression manquantes pour certains gènes dans certains échantillons. In the following fictive example, the gene Cyp2d22 has only one expression value, in GSM2545338 sample.
R
rna_with_missing_values <- rna %>%
select(gene, sample, expression) %>%
filter(gene %in% c("Asl", "Apod", "Cyp2d22")) %>%
filter(sample %in% c("GSM2545336", "GSM2545337", "GSM2545338")) %>%
arrange(sample) %>%
filter(!(gene == "Cyp2d22" & sample != "GSM2545338"))
rna_with_missing_values
SORTIE
# A tibble: 7 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Asl GSM2545337 361
4 Apod GSM2545337 10347
5 Asl GSM2545338 400
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545338 1603Par défaut, la fonction pivot_wider() ajoutera
NA pour les valeurs manquantes. Ceci peut être paramétré
avec l’argument values_fill de la fonction
pivot_wider().
R
rna_with_missing_values %>%
pivot_wider(names_from = échantillon,
values_from = expression)
ERREUR
Error in `pivot_wider()`:
! Can't select columns that don't exist.
✖ Column `échantillon` doesn't exist.R
rna_with_missing_values %>%
pivot_wider(names_from = échantillon,
values_from = expression,
valeurs_fill = 0)
ERREUR
Error in `pivot_wider()`:
! Can't select columns that don't exist.
✖ Column `échantillon` doesn't exist.Pivoter les données dans un format plus long
Dans la situation inverse, nous utilisons les noms de colonnes et les transformons en une paire de nouvelles variables. Une variable représente les noms de colonnes sous forme de valeurs , et l’autre variable contient les valeurs précédemment associées aux noms de colonnes.
pivot_longer() prend quatre arguments principaux :
- les données à transformer ;
- le
names_to: le nouveau nom de colonne que nous souhaitons créer et remplir avec les noms de colonnes actuels ; - les
values_to: le nouveau nom de colonne que nous souhaitons créer et remplir avec valeurs actuelles ; - les noms des colonnes à utiliser pour renseigner les variables
names_toetvalues_to(ou à supprimer).

rna.
To recreate rna_long from rna_wide we would
create a key called sample and value called
expression and use all columns except gene for
the key variable. Here we drop gene column with a minus
sign.
Notice how the new variable names are to be quoted here.
R
rna_long <- rna_wide %>%
pivot_longer(names_to = "sample",
values_to = "expression",
-gene)
rna_long
SORTIE
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Asl GSM2545339 586
5 Asl GSM2545340 626
6 Asl GSM2545341 988
7 Asl GSM2545342 836
8 Asl GSM2545343 535
9 Asl GSM2545344 586
10 Asl GSM2545345 597
# ℹ 32,418 more rowsNous aurions également pu utiliser une spécification indiquant les
colonnes à inclure. Cela peut être utile si vous disposez d’un grand
nombre de colonnes d’identification , et il est plus facile de spécifier
ce qu’il faut rassembler que ce qu’il faut laisser seul. Ici, la
fonction starts_with() peut aider à récupérer des exemples
de noms sans avoir à tous les lister ! Une autre possibilité serait
d’utiliser l’opérateur : !
R
rna_wide %>%
pivot_longer(names_to = "sample",
values_to = "expression",
cols = start_with("GSM"))
ERREUR
Error in `pivot_longer()`:
ℹ In argument: `start_with("GSM")`.
Caused by error in `start_with()`:
! could not find function "start_with"R
rna_wide %> %
pivot_longer(names_to = "échantillon",
valeurs_to = "expression",
GSM2545336:GSM2545380)
ERREUR
Error in rna_wide %> % pivot_longer(names_to = "échantillon", valeurs_to = "expression", : could not find function "%> %"Notez que si nous avions des valeurs manquantes dans le format large, le « NA » serait inclus dans le nouveau format long.
Souvenez-vous de notre précédent tibble fictif contenant des valeurs manquantes :
R
rna_with_missing_values
SORTIE
# A tibble: 7 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Asl GSM2545337 361
4 Apod GSM2545337 10347
5 Asl GSM2545338 400
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545338 1603R
wide_with_NA <- rna_with_missing_values %>%
pivot_wider(names_from = sample,
values_from = expression)
wide_with_NA
SORTIE
# A tibble: 3 × 4
gene GSM2545336 GSM2545337 GSM2545338
<chr> <dbl> <dbl> <dbl>
1 Asl 1170 361 400
2 Apod 36194 10347 9173
3 Cyp2d22 NA NA 1603R
wide_with_NA %>%
pivot_longer(names_to = "sample",
values_to = "expression",
-gene)
SORTIE
# A tibble: 9 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Apod GSM2545336 36194
5 Apod GSM2545337 10347
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545336 NA
8 Cyp2d22 GSM2545337 NA
9 Cyp2d22 GSM2545338 1603Passer à des formats plus larges et plus longs peut être un moyen utile d’équilibrer un ensemble de données afin que chaque réplique ait la même composition.
R
rna1 <- rna %>%
select(gène, souris, expression) %>%
pivot_wider(names_from = souris, valeurs_from = expression)
ERREUR
Error in `select()`:
! Can't select columns that don't exist.
✖ Column `gène` doesn't exist.R
rna1
ERREUR
Error in eval(expr, envir, enclos): object 'rna1' not foundR
rna1 %>%
pivot_longer(names_to = "mouse_id", valeurs_to = "counts", -gene)
ERREUR
Error in eval(expr, envir, enclos): object 'rna1' not foundERREUR
Error in `pivot_longer()`:
! Arguments in `...` must be used.
✖ Problematic argument:
• valeurs_to = "counts"
ℹ Did you misspell an argument name?Question
Sous-ensemble de gènes situés sur les chromosomes X et Y de la trame de données « rna » et répartissent la trame de données avec « sexe » en colonnes, « nom_chromosome » en lignes et l’expression moyenne des gènes localisés dans chaque chromosome comme valeurs, comme dans le tableau suivant :
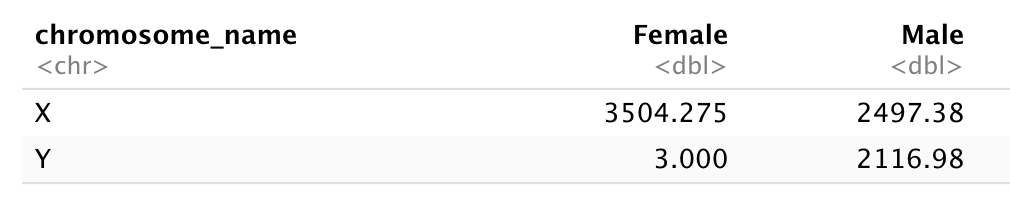
Il faudra résumer avant de remodeler !
Calculons d’abord le niveau d’expression moyen des gènes liés X et Y à partir de échantillons mâles et femelles…
R
arn %>%
filter(chromosome_name == "Y" | chromosome_name == "X") %>%
group_by(sex, chromosome_name) %>%
résumé(moyenne = moyenne(expression))
ERREUR
Error in résumé(., moyenne = moyenne(expression)): could not find function "résumé"Et faites pivoter le tableau au format large
R
rna_1 <- rna %>%
filter(chromosome_name == "Y" | chromosome_name == "X") %>%
group_by(sex, chromosome_name) %>%
summarise(mean = moyenne(expression)) %>%
pivot_wider(names_from = sexe,
valeurs_from = moyenne)
ERREUR
Error in `summarise()`:
ℹ In argument: `mean = moyenne(expression)`.
ℹ In group 1: `sex = "Female"` and `chromosome_name = "X"`.
Caused by error in `moyenne()`:
! could not find function "moyenne"R
rna_1
ERREUR
Error in eval(expr, envir, enclos): object 'rna_1' not foundMaintenant, prenez cette trame de données et transformez-la avec
pivot_longer() afin que chaque ligne soit une combinaison
unique de chromosome_name par gender.
R
rna_1 %>%
pivot_longer(names_to = "gender",
valeurs_to = "mean",
-chromosome_name)
ERREUR
Error in eval(expr, envir, enclos): object 'rna_1' not foundERREUR
Error in `pivot_longer()`:
! Arguments in `...` must be used.
✖ Problematic argument:
• valeurs_to = "mean"
ℹ Did you misspell an argument name?Calculons d’abord l’expression moyenne par gène et par temps
R
arn %>%
group_by(gène, temps) %>%
résumé(mean_exp = moyenne(expression))
ERREUR
Error in résumé(., mean_exp = moyenne(expression)): could not find function "résumé"avant d’utiliser la fonction pivot_wider()
R
rna_time <- rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.R
rna_time
SORTIE
# A tibble: 1,474 × 4
# Groups: gene [1,474]
gene `0` `4` `8`
<chr> <dbl> <dbl> <dbl>
1 AI504432 1034. 1104. 1014
2 AW046200 155. 152. 81
3 AW551984 238 302. 342.
4 Aamp 4603. 4870 4763.
5 Abca12 5.29 4.25 4.14
6 Abcc8 2576. 2609. 2292.
7 Abhd14a 591. 547. 432.
8 Abi2 4881. 4903. 4945.
9 Abi3bp 1175. 1061. 762.
10 Abl2 2170. 2078. 2131.
# ℹ 1,464 more rowsNotez que cela génère un tibble avec certains noms de colonnes commençant par un nombre. Si nous voulions sélectionner la colonne correspondant aux points temporels, nous ne pourrions pas utiliser directement les noms de colonnes… Que se passe-t-il lorsque l’on sélectionne la colonne 4 ?
R
rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
select(gene, 4)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene `8`
<chr> <dbl>
1 AI504432 1014
2 AW046200 81
3 AW551984 342.
4 Aamp 4763.
5 Abca12 4.14
6 Abcc8 2292.
7 Abhd14a 432.
8 Abi2 4945.
9 Abi3bp 762.
10 Abl2 2131.
# ℹ 1,464 more rowsPour sélectionner le timepoint 4, il faudrait citer le nom de la colonne, avec des backticks “\`”
R
rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
select(gene, `4`)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene `4`
<chr> <dbl>
1 AI504432 1104.
2 AW046200 152.
3 AW551984 302.
4 Aamp 4870
5 Abca12 4.25
6 Abcc8 2609.
7 Abhd14a 547.
8 Abi2 4903.
9 Abi3bp 1061.
10 Abl2 2078.
# ℹ 1,464 more rowsUne autre possibilité serait de renommer la colonne, en choisissant un nom qui ne commence pas par un chiffre :
R
rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
rename("time0" = `0`, "time4" = `4`, "time8" = `8`) %>%
select(gene, time4)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene time4
<chr> <dbl>
1 AI504432 1104.
2 AW046200 152.
3 AW551984 302.
4 Aamp 4870
5 Abca12 4.25
6 Abcc8 2609.
7 Abhd14a 547.
8 Abi2 4903.
9 Abi3bp 1061.
10 Abl2 2078.
# ℹ 1,464 more rowsQuestion
Utilisez la trame de données précédente contenant les niveaux d’expression moyens par point temporel et créez une nouvelle colonne contenant les changements de pli entre le point temporel 8 et le point temporel 0, et les changements de pli entre le point temporel 8 et le point temporel 4. Convertissez ce tableau en un tableau au format long regroupant les changements de pli calculés.
À partir du tibble rna_time :
R
arn_time
ERREUR
Error in eval(expr, envir, enclos): object 'arn_time' not foundCalculer les changements de plis :
R
rna_time %>%
muter (time_8_vs_0 = `8` / `0`, time_8_vs_4 = `8` / `4`)
ERREUR
Error in muter(., time_8_vs_0 = `8`/`0`, time_8_vs_4 = `8`/`4`): could not find function "muter"Et utilisez la fonction pivot_longer() :
R
rna_time %>%
mutate(time_8_vs_0 = `8` / `0`, time_8_vs_4 = `8` / `4`) %>%
pivot_longer(names_to = "comparisons",
values_to = "Fold_changes",
time_8_vs_0:time_8_vs_4)
SORTIE
# A tibble: 2,948 × 6
# Groups: gene [1,474]
gene `0` `4` `8` comparisons Fold_changes
<chr> <dbl> <dbl> <dbl> <chr> <dbl>
1 AI504432 1034. 1104. 1014 time_8_vs_0 0.981
2 AI504432 1034. 1104. 1014 time_8_vs_4 0.918
3 AW046200 155. 152. 81 time_8_vs_0 0.522
4 AW046200 155. 152. 81 time_8_vs_4 0.532
5 AW551984 238 302. 342. time_8_vs_0 1.44
6 AW551984 238 302. 342. time_8_vs_4 1.13
7 Aamp 4603. 4870 4763. time_8_vs_0 1.03
8 Aamp 4603. 4870 4763. time_8_vs_4 0.978
9 Abca12 5.29 4.25 4.14 time_8_vs_0 0.784
10 Abca12 5.29 4.25 4.14 time_8_vs_4 0.975
# ℹ 2,938 more rowsJoindre des tables
Dans de nombreuses situations réelles, les données sont réparties sur plusieurs tables. Cela se produit généralement parce que différents types d’informations sont collectés à partir de différentes sources.
Il peut être souhaitable que certaines analyses combinent les données de deux ou plusieurs tables en une seule trame de données basée sur une colonne qui serait commune à toutes les tables.
Le package dplyr fournit un ensemble de fonctions de
jointure pour combiner deux trames de données basées sur des
correspondances dans des colonnes spécifiées. Ici, nous fournissons une
brève introduction aux jointures. Pour en savoir plus, veuillez vous
référer au chapitre sur les jointures de table . La Data Transformation
Cheat Sheet fournit également un bref aperçu sur les jointures de
table.
Nous allons illustrer la jointure en utilisant une petite table,
rna_mini que nous allons créer en sous-définissant la table
rna d’origine, en ne gardant que 3 colonnes et 10
lignes.
R
rna_mini <- rna %>%
select(gène, échantillon, expression) %>%
head(10)
ERREUR
Error in `select()`:
! Can't select columns that don't exist.
✖ Column `gène` doesn't exist.R
rna_mini
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundLe deuxième tableau, annot1, contient 2 colonnes, gene
et gene_description. Vous pouvez soit télécharger
annot1.csv en cliquant sur le lien puis en vous déplaçant dans le
dossier data/, ou vous pouvez utiliser le code R ci-dessous
pour le télécharger directement dans le dossier.
R
download.file(url = "https://carpentries-incubator.github.io/bioc-intro/data/annot1.csv",
destfile = "data/annot1.csv")
annot1 <- read_csv(file = "data/annot1.csv")
annot1
SORTIE
# A tibble: 10 × 2
gene gene_description
<chr> <chr>
1 Cyp2d22 cytochrome P450, family 2, subfamily d, polypeptide 22 [Source:MGI S…
2 Klk6 kallikrein related-peptidase 6 [Source:MGI Symbol;Acc:MGI:1343166]
3 Fcrls Fc receptor-like S, scavenger receptor [Source:MGI Symbol;Acc:MGI:19…
4 Plp1 proteolipid protein (myelin) 1 [Source:MGI Symbol;Acc:MGI:97623]
5 Exd2 exonuclease 3'-5' domain containing 2 [Source:MGI Symbol;Acc:MGI:192…
6 Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88056]
7 Gnb4 guanine nucleotide binding protein (G protein), beta 4 [Source:MGI S…
8 Slc2a4 solute carrier family 2 (facilitated glucose transporter), member 4 …
9 Asl argininosuccinate lyase [Source:MGI Symbol;Acc:MGI:88084]
10 Gjc2 gap junction protein, gamma 2 [Source:MGI Symbol;Acc:MGI:2153060] Nous voulons maintenant joindre ces deux tables en une seule
contenant toutes les variables en utilisant la fonction
full_join() du package dplyr. La fonction
trouvera automatiquement la variable commune correspondant aux colonnes
de la première et de la deuxième table. Dans ce cas, « gène » est la
variable commune . Ces variables sont appelées clés. Les clés sont
utilisées pour faire correspondre observations dans différentes
tables.
R
full_join(rna_mini, annot1)
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundDans la vraie vie, les annotations génétiques sont parfois étiquetées différemment.
La table annot2 est exactement la même que
annot1 sauf que la variable contenant les noms de gènes est
étiquetée différemment. Encore une fois, soit télécharger
annot2.csv vous-même et déplacez-le vers data/ ou
utilisez le code R ci-dessous.
R
download.file(url = "https://carpentries-incubator.github.io/bioc-intro/data/annot2.csv",
destfile = "data/annot2.csv")
annot2 <- read_csv(file = "data/annot2.csv")
annot2
SORTIE
# A tibble: 10 × 2
external_gene_name description
<chr> <chr>
1 Cyp2d22 cytochrome P450, family 2, subfamily d, polypeptide 22 [S…
2 Klk6 kallikrein related-peptidase 6 [Source:MGI Symbol;Acc:MGI…
3 Fcrls Fc receptor-like S, scavenger receptor [Source:MGI Symbol…
4 Plp1 proteolipid protein (myelin) 1 [Source:MGI Symbol;Acc:MGI…
5 Exd2 exonuclease 3'-5' domain containing 2 [Source:MGI Symbol;…
6 Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88056]
7 Gnb4 guanine nucleotide binding protein (G protein), beta 4 [S…
8 Slc2a4 solute carrier family 2 (facilitated glucose transporter)…
9 Asl argininosuccinate lyase [Source:MGI Symbol;Acc:MGI:88084]
10 Gjc2 gap junction protein, gamma 2 [Source:MGI Symbol;Acc:MGI:…Si aucun des noms de variables ne correspond, nous pouvons définir
manuellement les variables à utiliser pour la correspondance. Ces
variables peuvent être définies en utilisant l’argument by,
comme indiqué ci-dessous avec les tables rna_mini et
annot2.
R
full_join(rna_mini, annot2, by = c("gene" = "external_gene_name"))
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundComme on peut le voir ci-dessus, le nom de variable de la première table est conservé dans celle jointe.
Défi:
Téléchargez la table annot3 en cliquant sur ici
et placez la table dans votre Dépôt de données. À l’aide de la fonction
full_join() , joignez les tables rna_mini et
annot3. Que s’est-il passé pour les gènes Klk6,
mt-Tf, mt-Rnr1, mt-Tv, mt-Rnr2 et
mt-Tl1 ?
R
annot3 <- read_csv("data/annot3.csv")
full_join(rna_mini, annot3)
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundLes gènes Klk6 ne sont présents que dans
rna_mini, tandis que les gènes mt-Tf,
mt-Rnr1, mt-Tv, mt-Rnr2 et mt-Tl1
sont présent uniquement dans la table annot3. Leurs valeurs
respectives pour les variables du tableau ont été codées comme
manquantes.
Exporter des données
Maintenant que vous avez appris à utiliser dplyr pour
extraire des informations de ou résumer vos données brutes, vous
souhaiterez peut-être exporter ces nouveaux ensembles de données pour
les partager avec vos collaborateurs ou pour les archiver.
Semblable à la fonction read_csv() utilisée pour lire
les fichiers CSV dans R, il existe une fonction write_csv()
qui génère des fichiers CSV à partir de trames de données.
Avant d’utiliser write_csv(), nous allons créer un
nouveau dossier, data_output, dans notre répertoire de
travail qui stockera cet ensemble de données généré. Nous ne voulons pas
que écrive les ensembles de données générés dans le même répertoire que
nos données brutes. C’est une bonne pratique de les garder séparés. Le
dossier data ne doit contenir que les données brutes et non
modifiées, et doit être laissé seul pour nous assurer que nous ne
supprimons pas ou ne le modifions pas. En revanche, notre script
générera le contenu du répertoire data_output , donc même
si les fichiers qu’il contient sont supprimés, nous pouvons toujours les
régénérer.
Utilisons write_csv() pour sauvegarder la table rna_wide
que nous avons créée précédemment.
R
write_csv(rna_wide, file = "data_output/rna_wide.csv")