Content from Organisation des données avec des feuilles de calcul
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 60 minutes
Vue d'ensemble
Questions
- Comment organiser des données tabulaires ?
Objectifs
- Découvrez les feuilles de calcul, leurs forces et leurs faiblesses.
- Comment formater les données dans des feuilles de calcul pour une utilisation efficace des données ?
- Découvrez les erreurs courantes des feuilles de calcul et comment les corriger.
- Organisez vos données selon des principes de données propres.
- Découvrez les formats de feuilles de calcul textuels tels que les formats séparés par des virgules (CSV) ou par des tabulations (TSV).
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Tableurs
Question
- Quels sont les principes de base d’utilisation des feuilles de calcul pour une bonne organisation des données ?
Objectifs
- Décrire les bonnes pratiques pour organiser les données afin que les ordinateurs puissent en faire la meilleure utilisation.
Point clé
- Une bonne organisation des données est la base de tout projet de recherche.
Une bonne organisation des données est la base de votre projet de recherche . La plupart des chercheurs disposent de données ou effectuent la saisie de données dans des feuilles de calcul . Les tableurs sont des interfaces graphiques très utiles pour concevoir des tableaux de données et gérer des fonctions de contrôle qualité de données très basiques. Voir aussi @Broman : 2018.
Aperçu de la feuille de calcul
Les feuilles de calcul sont utiles pour la saisie de données. Nous avons donc beaucoup de données dans des feuilles de calcul. Une grande partie de votre temps en tant que chercheur sera consacrée à cette étape de « gestion des données ». Ce n’est pas le plus amusant, mais c’est nécessaire. Nous vous apprendrons comment réfléchir à l’organisation des données et quelques pratiques pour une gestion plus efficace des données.
Ce que cette leçon ne vous apprendra pas
- Comment faire des statistiques dans une feuille de calcul
- Comment faire un traçage dans une feuille de calcul
- Comment écrire du code dans des tableurs
Si vous cherchez à faire cela, une bonne référence est Head First Excel, publié par O’Reilly.
Pourquoi n’enseignons-nous pas l’analyse des données dans des feuilles de calcul
L’analyse des données dans des feuilles de calcul nécessite généralement beaucoup de travail manuel. Si vous souhaitez modifier un paramètre ou exécuter une analyse avec un nouvel ensemble de données , vous devez généralement tout refaire à la main. (Nous savons que vous pouvez créer des macros, mais voyez le point suivant.)
Il est également difficile de suivre ou de reproduire des analyses statistiques ou graphiques effectuées dans des tableurs lorsque vous souhaitez revenir à votre travail ou que quelqu’un vous demande des détails sur votre analyse.
De nombreux tableurs sont disponibles. Étant donné que la plupart des participants utilisent Excel comme tableur principal, cette leçon utilisera des exemples Excel. Un tableur gratuit qui peut également être utilisé est LibreOffice. Les commandes peuvent différer un peu selon les programmes, mais l’idée générale est la même.
Les tableurs englobent de nombreuses choses que nous devons pouvoir faire en tant que chercheurs. Nous pouvons les utiliser pour :
- La saisie des données
- Organisation des données
- Sous-ensemble et tri des données
- Statistiques
- Traçage
Les tableurs utilisent des tableaux pour représenter et afficher les données. Les données formatées sous forme de tableaux sont également le thème principal de ce chapitre, et nous verrons comment organiser les données en tableaux de manière standardisée pour assurer une analyse efficace en aval.
Défi : Discutez des points suivants avec votre voisin
- Avez-vous utilisé des feuilles de calcul, dans vos recherches, vos cours, ou à la maison ?
- Quel type d’opérations effectuez-vous dans des feuilles de calcul ?
- Selon vous, pour lesquels les feuilles de calcul sont-elles utiles ?
- Avez-vous accidentellement fait quelque chose dans un tableur qui vous a rendu frustré ou triste ?
Problèmes avec les feuilles de calcul
Les feuilles de calcul sont utiles pour la saisie de données, mais en réalité, nous avons tendance à utiliser des tableurs pour bien plus que la saisie de données. Nous les utilisons pour créer des tableaux de données pour les publications, pour générer des statistiques récapitulatives et réaliser des chiffres.
Générer des tableaux pour des publications dans une feuille de calcul n’est pas optimal - souvent, lors du formatage d’un tableau de données, nous rapportons les principales statistiques récapitulatives d’une manière qui n’est pas vraiment destinée à être lue comme des données, et implique souvent un formatage spécial (fusion de cellules, création de bordures, préférences esthétiques pour les couleurs, etc.). Nous vous conseillons de effectuer ce genre d’opération au sein de votre logiciel d’édition de documents.
Ces deux dernières applications, génératrices de statistiques et de chiffres, doivent être utilisées avec précaution : en raison de la nature graphique, par glisser-déposer des tableurs, il peut être très difficile, voire impossible, de reproduisez vos pas (et encore moins retracer ceux de quelqu’un d’autre), en particulier si vos statistiques ou chiffres nécessitent que vous fassiez des calculs plus complexes. De plus, en effectuant des calculs dans une feuille de calcul, il est facile d’appliquer accidentellement une formule légèrement différente à plusieurs cellules adjacentes. Lorsque vous utilisez un programme de statistiques basé sur une ligne de commande comme R ou SAS, il est pratiquement impossible d’appliquer un calcul à une observation de votre ensemble de données mais pas à une autre, sauf si vous le faites cela exprès.
Utiliser des feuilles de calcul pour la saisie et le nettoyage des données
Dans cette leçon, nous supposerons que vous utilisez très probablement Excel comme votre tableur principal - il en existe d’autres (gnumeric, Calc d’OpenOffice), et leurs fonctionnalités sont similaires, mais Excel semble est le programme le plus utilisé par les biologistes et les chercheurs biomédicaux.
Dans cette leçon, nous allons parler de :
- Formatage des tableaux de données dans des feuilles de calcul
- Problèmes de formatage
- Exporter des données
Formatage des tableaux de données dans des feuilles de calcul
Des questions
- Comment formater les données dans des feuilles de calcul pour une utilisation efficace des données ?
Objectifs
Décrire les meilleures pratiques pour la saisie et le formatage des données dans les feuilles de calcul .
Appliquez les meilleures pratiques pour organiser les variables et les observations dans une feuille de calcul .
Points clés
Ne modifiez jamais vos données brutes. Faites toujours une copie avant d’apporter des modifications.
Gardez une trace de toutes les étapes que vous suivez pour nettoyer vos données dans un fichier texte brut .
Organisez vos données selon des principes de données ordonnés.
L’erreur la plus courante est de traiter les tableurs comme des cahiers de laboratoire , c’est-à-dire de s’appuyer sur le contexte, les notes dans la marge, la disposition spatiale des données et des champs pour transmettre des informations. En tant qu’humains, nous pouvons (généralement) interpréter ces choses, mais les ordinateurs ne voient pas les informations de la même manière, et à moins que nous expliquions à l’ordinateur ce que chaque signifie (et ça peut être dur !), il ne pourra pas voir comment nos données s’emboîtent.
En utilisant la puissance des ordinateurs, nous pouvons gérer et analyser les données de manière beaucoup plus efficace et plus rapide, mais pour utiliser cette puissance, nous devons configurer nos données pour que l’ordinateur puisse comprenez-le (et les ordinateurs sont très littéraux).
C’est pourquoi il est extrêmement important de mettre en place des tableaux bien formatés dès le départ - avant même de commencer à saisir les données de votre toute première expérience préliminaire . L’organisation des données est le fondement de votre projet de recherche. Cela peut rendre plus facile ou plus difficile le travail avec vos données tout au long de votre analyse, il vaut donc la peine de réfléchir au moment où vous effectuez votre saisie de données ou configurez votre expérience. Vous pouvez configurer les choses de différentes manières dans des feuilles de calcul, mais certains de ces choix peuvent limiter votre capacité à travailler avec les données dans d’autres programmes ou vous empêcher de- Dans 6 mois ou votre collaborateur travaillera avec les données.
Remarque : les meilleures mises en page/formats (ainsi que les logiciels et les interfaces ) pour la saisie et l’analyse des données peuvent être différentes. Il est important d’en tenir compte, et idéalement d’automatiser la conversion de l’un à l’autre.
Garder une trace de vos analyses
Lorsque vous travaillez avec des feuilles de calcul, lors d’un nettoyage de données ou d’analyses , il est très facile de vous retrouver avec une feuille de calcul qui semble très différente de celle avec laquelle vous avez commencé. Afin de pouvoir reproduire vos analyses ou comprendre ce que vous avez fait lorsqu’un évaluateur ou un instructeur demande une analyse différente, vous devez
créez un nouveau fichier avec vos données nettoyées ou analysées. Ne modifiez pas l’ensemble de données d’origine, sinon vous ne saurez jamais par où vous avez commencé !
gardez une trace des étapes que vous avez suivies lors de votre nettoyage ou de votre analyse. Vous devez suivre ces étapes comme vous le feriez pour n’importe quelle étape d’une expérience. Nous vous recommandons de le faire dans un fichier texte brut stocké dans le même dossier que le fichier de données.
Ceci pourrait être un exemple de configuration de feuille de calcul :
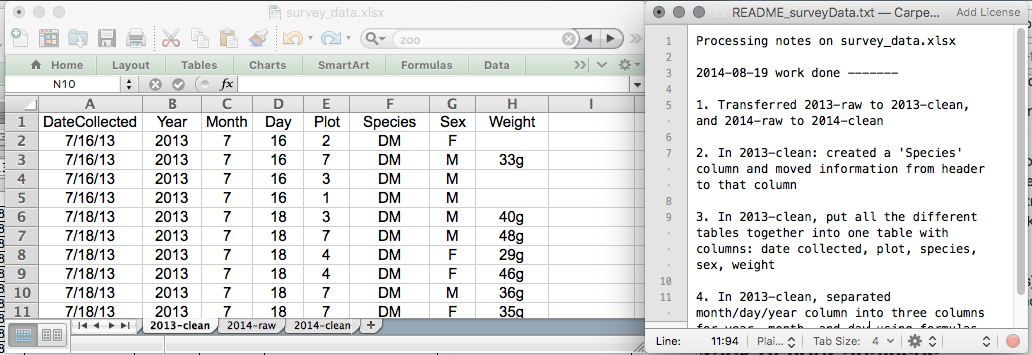
Mettez ces principes en pratique aujourd’hui lors de vos exercices.
Bien que la gestion des versions soit hors de portée de ce cours, vous pouvez consulter la leçon Menuiseries sur ‘Git’ pour découvrez comment maintenir le contrôle de version sur vos données. Voir aussi ce blog post pour un tutoriel rapide ou @Perez-Riverol:2016 pour une approche plus orientée recherche cas d’utilisation.
Structuration des données dans des feuilles de calcul
Les règles cardinales de l’utilisation des tableurs pour les données :
- Mettez toutes vos variables dans des colonnes - la chose que vous mesurez, comme « poids » ou « température ».
- Placez chaque observation dans sa propre rangée.
- Ne combinez pas plusieurs informations dans une seule cellule. Parfois cela semble être une chose, mais pensez que si c’est la seule façon vous voudrez pouvoir utiliser ou trier ces données.
- Laissez les données brutes brutes – ne les modifiez pas !
- Exportez les données nettoyées dans un format texte tel que le format CSV (valeurs séparées par des virgules). Cela garantit que n’importe qui peut utiliser les données et est requis par la plupart des référentiels de données.
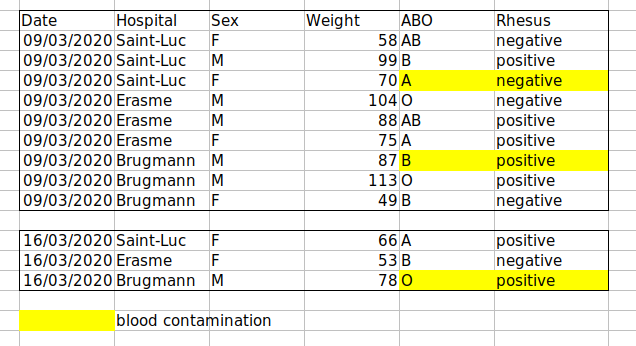
Par exemple, nous disposons de données provenant de patients ayant visité plusieurs hôpitaux à Bruxelles, en Belgique. Ils ont enregistré la date de la visite, l’hôpital, le sexe, le poids et le groupe sanguin des patients.
Si nous devions garder une trace des données comme ceci :
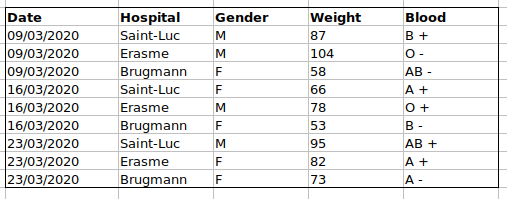
le problème est que les groupes ABO et Rhésus sont dans la même
colonne de type Blood . Donc, s’ils voulaient examiner
toutes les observations du groupe A ou examiner les distributions de
poids par groupe ABO, il serait difficile de le faire en utilisant cette
configuration de données. Si à la place nous mettions les groupes ABO et
Rhésus dans des colonnes différentes, vous voyez que ce serait beaucoup
plus facile.
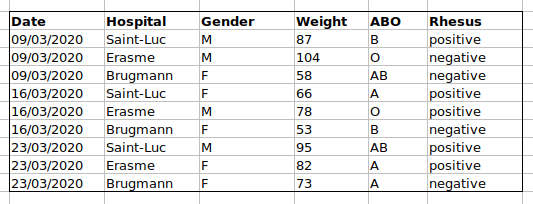
Une règle importante lors de la création d’une feuille de données est que les colonnes sont utilisées pour les variables et les lignes sont utilisées pour les observations :
- les colonnes sont des variables
- les lignes sont des observations
- les cellules sont des valeurs individuelles
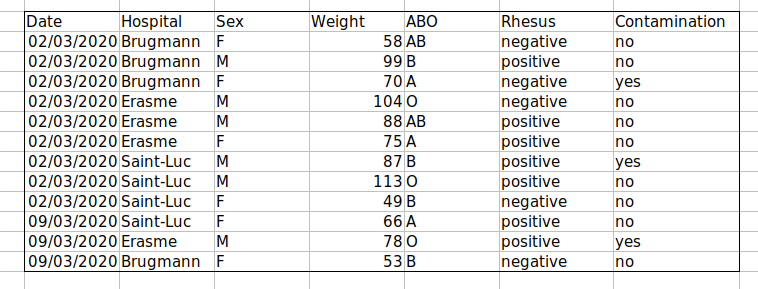
Défi : Nous allons prendre un ensemble de données désordonné et décrire comment nous allons le nettoyer.
Téléchargez un ensemble de données désordonné en cliquant sur ici.
Ouvrez les données dans un tableur.
Vous pouvez voir qu’il y a deux onglets. Les données contiennent diverses variables cliniques enregistrées dans divers hôpitaux bruxellois lors des première et deuxième vagues de COVID-19 en 2020. Comme vous pouvez le constater, les données ont été enregistrées différemment lors des vagues de mars et novembre. Vous êtes désormais la personne en charge de ce projet et vous souhaitez que puisse commencer à analyser les données.
Avec la personne à côté de vous, identifiez ce qui ne va pas avec cette feuille de calcul . Discutez également des étapes que vous devrez suivre pour nettoyer les onglets de la première et de la deuxième vague, et pour les rassembler tous dans une seule feuille de calcul .
Important : N’oubliez pas notre premier conseil : pour créer un nouveau fichier (ou onglet) pour les données nettoyées, ne modifiez jamais vos données (brutes) d’origine.
Après avoir effectué cet exercice, nous discuterons en groupe de ce qui n’allait pas avec ces données et de la manière dont vous pourriez y remédier.
Défi : Une fois que vous avez rangé les données, répondez aux questions suivantes :
- Combien d’hommes et de femmes ont participé à l’étude ?
- Combien de types A, AB et B ont été testés ?
- Comme ci-dessus, mais sans tenir compte des échantillons contaminés ?
- Combien de Rhésus + et - ont été testés ?
- Combien de donneurs universels (O-) ont été testés ?
- Quel est le poids moyen des hommes AB ?
- Combien d’échantillons ont été testés dans les différents hôpitaux ?
Une excellente référence, en particulier en ce qui concerne les scripts R est l’article Tidy Data @Wickham:2014.
Erreurs courantes dans les feuilles de calcul
Des questions
- Quels sont les défis courants liés au formatage des données dans les feuilles de calcul et comment pouvons-nous les éviter ?
Objectifs
- Reconnaître et résoudre les problèmes courants de formatage des feuilles de calcul.
Points clés
- Évitez d’utiliser plusieurs tableaux dans une même feuille de calcul.
- Évitez de répartir les données sur plusieurs onglets.
- Enregistrez les zéros comme des zéros.
- Utilisez une valeur nulle appropriée pour enregistrer les données manquantes.
- N’utilisez pas de formatage pour transmettre des informations ou pour donner une jolie apparence à votre feuille de calcul.
- Placez les commentaires dans une colonne séparée.
- Enregistrez les unités dans les en-têtes de colonnes.
- Incluez une seule information dans une cellule.
- Évitez les espaces, les chiffres et les caractères spéciaux dans les en-têtes de colonnes.
- Évitez les caractères spéciaux dans vos données.
- Enregistrez les métadonnées dans un fichier texte brut séparé.
Il y a quelques erreurs potentielles à surveiller dans vos propres données ainsi que dans les données de vos collaborateurs ou d’Internet. Si vous êtes conscient des erreurs et de l’effet négatif possible sur l’analyse des données en aval et l’interprétation des résultats, cela pourrait vous motiver ainsi que les membres de votre projet à essayer de les éviter. Apporter de petits changements à la façon dont vous formatez vos données dans des feuilles de calcul peut avoir un grand impact sur l’efficacité et la fiabilité en matière de nettoyage et d’analyse des données.
- Utiliser plusieurs tables
- Utiliser plusieurs onglets
- Ne pas remplir les zéros
- Utilisation de valeurs nulles problématiques
- Utiliser le formatage pour transmettre des informations
- Utiliser le formatage pour rendre la fiche technique jolie
- Placer des commentaires ou des unités dans des cellules
- Saisie de plusieurs informations dans une cellule
- Utilisation de noms de champs problématiques
- Utilisation de caractères spéciaux dans les données
- Inclusion de métadonnées dans le tableau de données
Utilisation de plusieurs tables
Une stratégie courante consiste à créer plusieurs tableaux de données dans une seule feuille de calcul . Cela perturbe l’ordinateur, alors ne faites pas ça ! Lorsque vous créez plusieurs tableaux dans une même feuille de calcul, vous établissez de fausses associations entre les éléments pour l’ordinateur, qui considère chaque ligne comme une observation. Vous utilisez également potentiellement le même nom de champ à plusieurs endroits, ce qui rendra plus difficile le nettoyage de vos données dans un formulaire utilisable. L’exemple ci-dessous illustre le problème :
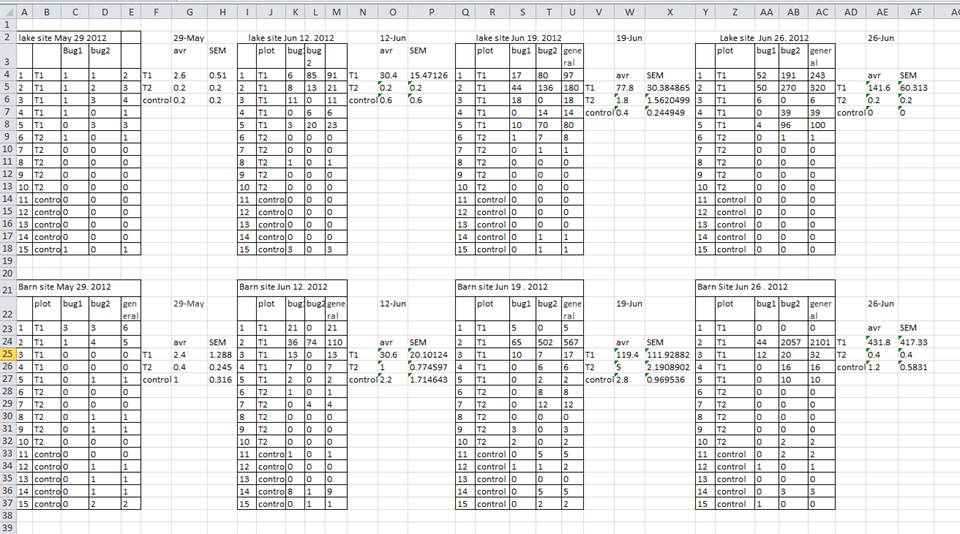
Dans l’exemple ci-dessus, l’ordinateur verra (par exemple) la ligne 4 et supposera que toutes les colonnes A-AF font référence au même échantillon. Cette ligne représente en fait quatre échantillons distincts (échantillon 1 pour chacune des quatre dates de collecte différentes - 29 mai, 12 juin, 19 juin et 26 juin), ainsi que quelques statistiques récapitulatives calculées (une moyenne (avr) et une erreur type de mesure (SEM)) pour deux de ces échantillons. D’autres lignes posent également problème.
Utiliser plusieurs onglets
Mais qu’en est-il des onglets du classeur ? Cela semble être un moyen simple d’organiser les données , n’est-ce pas ? Eh bien, oui et non. Lorsque vous créez des onglets supplémentaires, vous ne parvenez pas à permettre à l’ordinateur de voir les connexions dans les données qui s’y trouvent (vous devez introduire des fonctions spécifiques à l’application de feuille de calcul ou des scripts pour garantir cette connexion). Supposons, par exemple, que vous aillez créé un onglet séparé pour chaque jour où vous prenez une mesure.
Ce n’est pas une bonne pratique pour deux raisons :
vous êtes plus susceptible d’ajouter accidentellement des incohérences à vos données si à chaque fois que vous prenez une mesure, vous commencez à enregistrer les données dans un nouvel onglet, et
même si vous parvenez à empêcher toute incohérence, vous vous ajoutez une étape supplémentaire avant même d’analyser les données car vous devrez combiner ces données en une seule table de données. Vous devrez indiquer explicitement à l’ordinateur comment combiner les onglets - et si les onglets ne sont pas formatés de manière cohérente, vous devrez peut-être même le faire manuellement.
La prochaine fois que vous saisirez des données et que vous créerez un autre onglet ou un autre tableau, demandez-vous si vous pourriez éviter d’ajouter cet onglet en ajoutant une autre colonne à votre feuille de calcul d’origine. Nous avons utilisé plusieurs onglets dans notre exemple de fichier de données désordonné, mais vous avez maintenant vu comment vous pouvez réorganiser vos données pour les consolider entre les onglets.
Votre fiche technique peut devenir très longue au cours de l’expérience . Cela rend plus difficile la saisie des données si vous ne voyez pas vos en-têtes en haut de la feuille de calcul. Mais ne répétez pas votre ligne d’en-tête . Ceux-ci peuvent facilement être mélangés aux données, entraînant des problèmes plus tard. Au lieu de cela, vous pouvez geler les en-têtes de la colonne afin qu’ils restent visibles même lorsque vous disposez d’une feuille de calcul comportant plusieurs lignes.
Ne pas remplir les zéros
Il se peut que lorsque vous mesurez quelque chose, il s’agisse généralement d’un zéro, , par exemple le nombre de fois qu’un lapin est observé dans l’enquête. Pourquoi s’embêter à écrire le chiffre zéro dans cette colonne, alors qu’il s’agit principalement de zéros ?
Cependant, il existe une différence entre un zéro et une cellule vide dans une feuille de calcul . Pour l’ordinateur, un zéro est en réalité une donnée. Vous l’avez mesuré ou compté. Une cellule vide signifie qu’elle n’a pas été mesurée et l’ordinateur l’interprétera comme une valeur inconnue (également appelée valeur nulle ou valeur manquante).
Les feuilles de calcul ou les programmes statistiques interpréteront probablement mal les cellules vides que vous envisagez d’être des zéros. En n’entrant pas la valeur de votre observation, vous dites à votre ordinateur de représenter ces données comme inconnues ou manquantes (nulles). Cela peut entraîner des problèmes lors des calculs ou analyses ultérieurs. Par exemple, la moyenne d’un ensemble de nombres qui comprend une seule valeur nulle est toujours nulle (car l’ordinateur ne peut pas deviner la valeur des observations manquantes). Parce que de cela, il est très important d’enregistrer les zéros comme des zéros et vraiment les données manquantes comme des valeurs nulles.
Utilisation de valeurs nulles problématiques
Exemple : utiliser -999 ou d’autres valeurs numériques (ou zéro) pour représente les données manquantes.
Solutions:
Il existe plusieurs raisons pour lesquelles les valeurs nulles sont représentées différemment dans un ensemble de données. Parfois, des valeurs nulles déroutantes sont automatiquement enregistrées à partir de l’appareil de mesure. Si tel est le cas, vous ne pouvez pas faire grand-chose, mais cela peut être résolu lors du nettoyage des données avec un outil comme OpenRefine avant analyse. D’autres fois, différentes valeurs nulles sont utilisées pour transmettre différentes raisons pour lesquelles les données ne sont pas là. Il s’agit d’une information importante à capturer, mais elle utilise en fait une seule colonne pour capturer deux informations. Like for using formatting to convey information it would be good here to create a new column like ‘data_missing’ and use that column to capture the different reasons.
Quelle que soit la raison, c’est un problème si des données inconnues ou manquantes sont enregistrées comme -999, 999 ou 0.
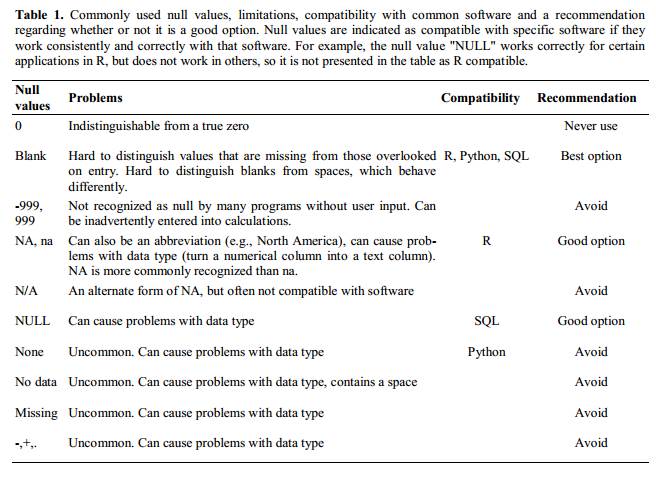
Many statistical programs will not recognise that these are intended to represent missing (null) values. La façon dont ces valeurs sont interprétées dépendra du logiciel que vous utilisez pour analyser vos données. Il est essentiel d’utiliser un indicateur nul clairement défini et cohérent.
Les blancs (la plupart des applications) et NA (pour R) sont de bons choix . @White : 2013 explique les bons choix pour indiquer des valeurs nulles pour différentes applications logicielles dans leur article :
Utiliser le formatage pour transmettre des informations
Exemple : mise en évidence des cellules, des lignes ou des colonnes qui doivent être exclues d’une analyse, en laissant des lignes vides pour indiquer séparations dans les données.
Solution : créez un nouveau champ pour coder les données qui doivent être exclues.
Utiliser le formatage pour rendre la fiche technique jolie {#formatting_pretty}
Exemple : fusion de cellules.
Solution : Si vous ne faites pas attention, le formatage d’une feuille de calcul pour qu’elle soit plus esthétiquement peut compromettre la capacité de votre ordinateur à voir les associations dans les données. Les cellules fusionnées rendront vos données illisibles par les logiciels de statistiques. Pensez à restructurer vos données de telle manière que vous n’aurez pas besoin de fusionner des cellules pour organiser vos données.
Placer des commentaires ou des unités dans des cellules
La plupart des logiciels d’analyse ne peuvent pas voir les commentaires Excel ou LibreOffice, et serait dérouté par les commentaires placés dans vos cellules de données. Comme décrit ci-dessus pour le formatage, créez un autre champ si vous devez ajouter des notes aux cellules. De même, n’incluez pas d’unités dans les cellules : idéalement, toutes les mesures que vous placez dans une colonne devraient être dans la même unité , mais si pour une raison quelconque ce n’est pas le cas, créez un autre champ et spécifient les unités dans lesquelles se trouve la cellule.
Saisir plusieurs informations dans une cellule
Exemple : Enregistrement des groupes ABO et Rhésus dans une seule cellule, tels que A+, B+, A-, …
Solution : N’incluez pas plus d’une information dans une cellule . Cela limitera les façons dont vous pourrez analyser vos données. Si vous avez besoin de ces deux mesures, concevez votre fiche technique pour inclure ces informations. Par exemple, incluez une colonne pour le groupe ABO et une pour le groupe Rhésus.
Utilisation de noms de champs problématiques {#field_name}
Choisissez des noms de champs descriptifs, mais veillez à ne pas inclure d’espaces, de chiffres ou de caractères spéciaux de quelque nature que ce soit. Les espaces peuvent être mal interprétés par les analyseurs qui utilisent des espaces comme délimiteurs et certains programmes n’aiment pas les noms de champs qui sont des chaînes de texte commençant par nombres.
Les traits de soulignement (_) sont une bonne
alternative aux espaces. Pensez à écrire les noms en casse chameau
(comme ceci : SampleFileName) pour améliorer la lisibilité de .
N’oubliez pas que les abréviations qui ont un sens pour le moment ne
seront peut-être pas si évidentes dans 6 mois, mais n’en faites pas trop
avec des noms qui sont excessivement longs. L’inclusion des unités dans
les noms de champs évite toute confusion et permet aux autres
d’interpréter facilement vos champs.
Exemples
| Réputation | Bonne alternative | Éviter |
|---|---|---|
| Max_temp_C | Température maximale | Température maximale (°C) |
| Précipitations_mm | Précipitation | précm |
| Moyenne_année_croissance | Croissance annuelle moyenne | Croissance moyenne/an |
| sexe | sexe | H/F |
| poids | poids | w. |
| cellule_type | Type de cellule | Type de cellule |
| Observation_01 | première_observation | 1er Obs. |
Utilisation de caractères spéciaux dans les données
Exemple : Vous traitez votre tableur comme un traitement de texte lorsque vous rédigez des notes, par exemple en copiant des données directement depuis Word ou d’autres applications.
Solution : Il s’agit d’une stratégie courante. Par exemple, lorsqu’ils écrivent un texte plus long dans une cellule, les utilisateurs incluent souvent des sauts de ligne, des tirets cadratins, , etc. dans leur feuille de calcul. De plus, lors de la copie de données à partir d’applications telles que Word, le formatage et les caractères non standard (tels que les guillemets alignés à gauche et à droite) sont inclus. Lors de l’exportation de ces données dans un environnement de codage/statistique ou dans une base de données relationnelle, des choses dangereuses peuvent se produire, comme des lignes coupées en deux et des erreurs d’encodage générées.
La meilleure pratique générale consiste à éviter d’ajouter des caractères tels que des nouvelles lignes, des tabulations et des tabulations verticales. In other words, treat a text cell as if it were a simple web form that can only contain text and spaces.
Inclusion de métadonnées dans le tableau de données
Exemple : Vous ajoutez une légende en haut ou en bas de votre tableau de données expliquant la signification des colonnes, les unités, les exceptions, etc.
Solution : L’enregistrement des données sur vos données (“métadonnées”) est essentiel. You may be on intimate terms with your dataset while you are collecting and analysing it, but the chances that you will still remember that the variable “sglmemgp” means single member of group, for example, or the exact algorithm you used to transform a variable or create a derived one, after a few months, a year, or more are slim.
De plus, il existe de nombreuses raisons pour lesquelles d’autres personnes pourraient vouloir examiner ou utiliser vos données : pour comprendre vos conclusions, pour vérifier vos conclusions, pour examiner la publication que vous avez soumise, pour reproduire vos résultats, pour concevoir une étude similaire, ou même archiver vos données pour y accéder et réutiliser par d’autres. Bien que les données numériques soient par définition lisibles par machine, comprendre leur signification est un travail pour les êtres humains. L’importance de documenter vos données pendant la phase de collecte et d’analyse de votre recherche ne peut être surestimée, surtout si votre recherche doit faire partie du dossier scientifique .
Cependant, les métadonnées ne doivent pas être contenues dans le fichier de données lui-même. Contrairement à un tableau dans un article ou un fichier supplémentaire, les métadonnées (sous sous forme de légendes) ne doivent pas être incluses dans un fichier de données puisque ces informations ne sont pas des données, et leur inclusion peut perturber la façon dont les programmes informatiques interprètent votre fichier de données. Les métadonnées doivent plutôt être stockées en tant que fichier distinct dans le même répertoire que votre fichier de données, de préférence au format texte brut avec un nom qui l’associe clairement à votre fichier de données. . Because metadata files are free text format, they also allow you to encode comments, units, information about how null values are encoded, etc. that are important to document but can disrupt the formatting of your data file.
De plus, les métadonnées au niveau du fichier ou de la base de données décrivent comment les fichiers qui constituent l’ensemble de données sont liés les uns aux autres ; dans quel format ils se trouvent ; et s’ils remplacent ou sont remplacés par les fichiers précédents. Un fichier readme.txt au niveau du dossier est la manière classique de comptabiliser tous les fichiers et dossiers d’un projet.
(Texte sur les métadonnées adapté du cours en ligne Research Data MANTRA par EDINA et Data Library, Université d’Édimbourg. MANTRA est sous licence Creative Commons Attribution 4.0 International Licence.)
Exporter des données
Question
- Comment pouvons-nous exporter des données à partir de feuilles de calcul d’une manière utile pour les applications en aval ?
Objectifs
- Stockez les données des feuilles de calcul dans des formats de fichiers universels.
- Exportez les données d’une feuille de calcul vers un fichier CSV.
Points clés
Les données stockées dans des formats de feuilles de calcul courants ne seront souvent pas lues correctement dans un logiciel d’analyse de données, introduisant des erreurs dans vos données .
L’exportation de données à partir de feuilles de calcul vers des formats tels que CSV ou TSV les place dans un format qui peut être utilisé de manière cohérente par la plupart des programmes.
Le stockage des données avec lesquelles vous allez travailler pour
vos analyses dans le format de fichier Excel par défaut
(*.xls ou *.xlsx - selon la version d’Excel )
n’est pas une bonne idée. Pourquoi?
Parce qu’il s’agit d’un format propriétaire, et qu’il est possible que dans le futur, la technologie n’existe pas (ou devienne suffisamment rare) pour rendre l’ouverture du fichier peu pratique, voire impossible. déposer.
D’autres logiciels de tableur peuvent ne pas être en mesure d’ouvrir les fichiers enregistrés dans un format Excel propriétaire .
Différentes versions d’Excel peuvent gérer les données différemment, entraînant des incohérences. Dates est un exemple bien documenté d’incohérences dans le stockage de données.
Enfin, de plus en plus de revues et d’organismes subventionnaires vous demandent de déposer vos données dans un référentiel de données, et la plupart d’entre elles n’acceptent pas le format Excel. Il doit être dans l’un des formats discutés ci-dessous.
Les points ci-dessus s’appliquent également à d’autres formats tels que les formats open data utilisés par LibreOffice / Open Office. Ces formats ne sont pas statiques et ne sont pas analysés de la même manière par différents packages logiciels .
Le stockage des données dans un format universel, ouvert et statique aidera à résoudre ce problème. Essayez les valeurs délimitées par des tabulations (valeurs séparées par des tabulations ou TSV) ou délimitées par des virgules (valeurs séparées par des virgules ou CSV). Les fichiers CSV sont des fichiers texte simples où les colonnes sont séparées par des virgules, d’où « valeurs séparées par des virgules » ou CSV. L’avantage d’un fichier CSV par rapport à un Excel/SPSS/etc. est que nous pouvons ouvrir et lire un fichier CSV en utilisant à peu près n’importe quel logiciel, y compris des éditeurs de texte brut comme TextEdit ou NotePad. Les données d’un fichier CSV peuvent également être facilement importées dans d’autres formats et environnements, tels que SQLite et R. Nous ne sommes pas liés à une certaine version d’un certain programme coûteux lorsque nous travaillons avec CSV fichiers, c’est donc un bon format avec lequel travailler pour une portabilité maximale et une endurance. La plupart des tableurs peuvent facilement enregistrer au format texte délimité comme CSV, bien qu’ils puissent vous avertir lors de l’exportation du fichier.
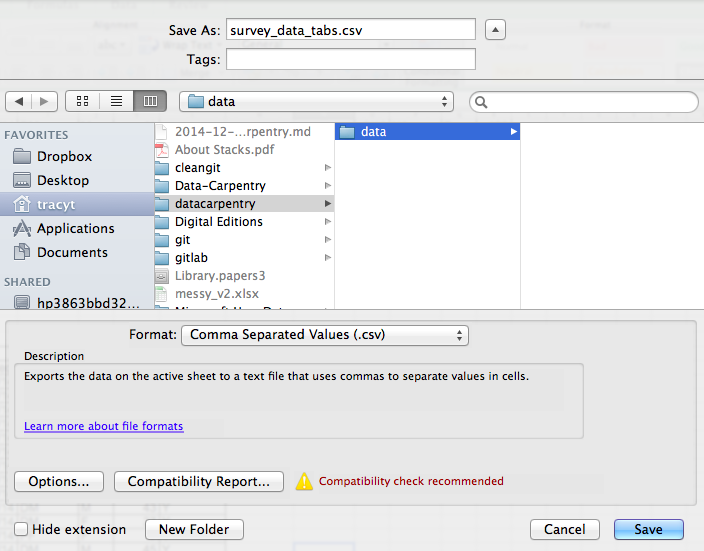
Pour enregistrer un fichier que vous avez ouvert dans Excel au format CSV :
- Dans le menu supérieur, sélectionnez « Fichier » et « Enregistrer sous ».
- Dans le champ « Format », dans la liste, sélectionnez « Valeurs
séparées par des virgules » (
*.csv). - Vérifiez le nom du fichier et l’emplacement où vous souhaitez l’enregistrer et cliquez sur « Enregistrer ».
Une remarque importante pour la rétrocompatibilité : vous pouvez ouvrir les fichiers CSV dans Excel !
Une note sur R et xls : Il existe des
packages R qui peuvent lire les fichiers xls (ainsi que les
feuilles de calcul Google). Il est même possible d’accéder à différentes
feuilles de calcul dans les documents xls.
Mais
- certains d’entre eux ne fonctionnent que sous Windows.
- cela équivaut à remplacer une exportation (simple mais manuelle)
vers
csvpar complexité/dépendances supplémentaires dans le code R d’analyse des données. - Les meilleures pratiques en matière de formatage des données s’appliquent toujours.
- Y a-t-il vraiment une bonne raison pour laquelle
csv(ou similaire) n’est pas adéquat ?
Mises en garde concernant les virgules
Dans certains ensembles de données, les valeurs des données elles-mêmes peuvent inclure des virgules (,). Dans ce cas, le logiciel que vous utilisez (y compris Excel) affichera très probablement de manière incorrecte les données en colonnes. En effet, les virgules qui font partie des valeurs de données seront interprétées comme des délimiteurs .
Par exemple, nos données pourraient ressembler à ceci :
species_id,genus,species,taxa
AB,Amphispiza,bilineata,Bird
AH,Ammospermophilus,harrisi,Rodent, not censused
AS,Ammodramus,savannarum,Bird
BA,Baiomys,taylori,RodentDans l’enregistrement « AH, Ammospermophilus, harrisi, Rongeur, non recensé », la valeur pour « taxons » comprend une virgule (« Rongeur, non recensé »). Si nous essayons de lire ce qui précède dans Excel (ou un autre tableur), nous obtiendrons quelque chose comme ceci :
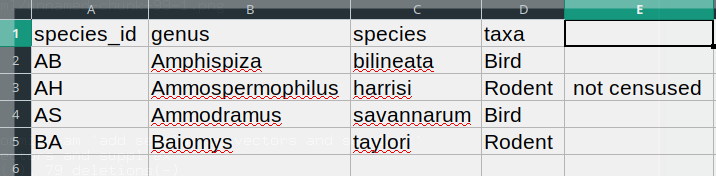
La valeur de « taxons » a été divisée en deux colonnes (au lieu d’être placée dans une seule colonne « D »). Cela peut se propager à un certain nombre d’autres erreurs . Par exemple, la colonne supplémentaire sera interprétée comme une colonne avec de nombreuses valeurs manquantes (et sans en-tête approprié). En plus de cela, la valeur dans la colonne « D » pour l’enregistrement de la ligne 3 (donc celle où la valeur de « taxons » contenait la virgule) est désormais incorrecte.
Si vous souhaitez stocker vos données au format csv et
vous attendez à ce que vos valeurs de données contiennent des virgules,
vous pouvez éviter le problème évoqué ci-dessus en mettant les valeurs
entre guillemets (““). En appliquant cette règle, nos données pourraient
ressembler à ceci :
species_id,genus,species,taxa
"AB","Amphispiza","bilineata","Bird"
"AH","Ammospermophilus","harrisi","Rodent, not censused"
"AS","Ammodramus","savannarum","Bird"
"BA","Baiomys","taylori","Rodent"Désormais, l’ouverture de ce fichier en tant que « csv » dans Excel n’entraînera pas une colonne supplémentaire, car Excel n’utilisera que des virgules qui se trouvent en dehors des guillemets comme caractères de délimitation.
Alternativement, si vous travaillez avec des données contenant des virgules, vous devrez probablement utiliser un autre délimiteur lorsque vous travaillerez dans une feuille de calcul 1. Dans ce cas, pensez à utiliser des tabulations comme délimiteur et à travailler avec des fichiers TSV. Les fichiers TSV peuvent être exportés à partir de feuilles de calcul de la même manière que les fichiers CSV.
Si vous travaillez avec un ensemble de données déjà existant dans lequel les valeurs de données ne sont pas incluses entre “” mais qui ont à la fois des virgules comme délimiteurs et des parties de valeurs de données, vous êtes potentiellement confronté à un problème majeur. avec nettoyage des données. Si l’ensemble de données que vous traitez contient des centaines ou des milliers d’enregistrements, nettoyez-les manuellement (soit en supprimant les virgules des valeurs de données, soit en mettant les valeurs entre guillemets - ““) non seulement va prendre des heures et des heures, mais peut finir par vous amener à introduire accidentellement de nombreuses erreurs.
Le nettoyage des ensembles de données est l’un des problèmes majeurs dans de nombreuses disciplines scientifiques . L’approche dépend presque toujours du contexte particulier. Cependant, il est recommandé de nettoyer les données de manière automatisée, par exemple en écrivant et en exécutant un script. Les leçons Python et R vous donneront les bases pour développer des compétences permettant de créer des scripts pertinents.
Résumé

Un flux de travail typique d’analyse de données est illustré dans la figure ci-dessus, où les données sont transformées, visualisées et modélisées à plusieurs reprises. Cette itération est répétée plusieurs fois jusqu’à ce que les données soient comprises. Cependant, dans de nombreux cas réels, la plupart du temps est consacré au nettoyage et à la préparation des données, plutôt qu’à leur analyse et à leur compréhension .
An agile data analysis workflow, with several fast iterations of the transform/visualise/model cycle is only feasible if the data is formatted in a predictable way and one can reason about the data without having to look at it and/or fix it.
Ceci est particulièrement pertinent dans les pays européens où la virgule est utilisée comme séparateur décimal . Dans de tels cas, le séparateur de valeurs par défaut dans un fichier csv sera le point-virgule (;), ou les valeurs seront systématiquement entre guillemets.↩︎
Content from R et RStudio
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 30 minutes
Vue d'ensemble
Questions
- Que sont R et RStudio ?
Objectifs
- Décrivez l’objectif des volets RStudio Script, Console, Environment et Plots.
- Organisez les fichiers et les répertoires pour un ensemble d’analyses en tant que projet R et comprenez le but du répertoire de travail.
- Utilisez l’interface d’aide intégrée de RStudio pour rechercher plus d’informations sur les fonctions R.
- Montrez comment fournir suffisamment d’informations pour le dépannage avec la communauté des utilisateurs R.
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Qu’est-ce que R ? Qu’est-ce que RStudio ?
Le terme R est utilisé pour désigner le langage de programmation, l’environnement de calcul statistique et le logiciel qui interprète les scripts écrits à l’aide de celui-ci.
RStudio est actuellement un moyen très populaire non seulement d’écrire vos scripts R mais aussi d’interagir avec le logiciel R 1. Pour fonctionner correctement, RStudio a besoin de R et donc les deux doivent être installés sur votre ordinateur.
La RStudio IDE Cheat Sheet fournit beaucoup plus d’informations que ce qui sera couvert ici, mais peut être utile pour apprendre les raccourcis clavier et découvrir de nouvelles fonctionnalités.
Pourquoi apprendre R ?
R n’implique pas beaucoup de pointage et de clic, et c’est une bonne chose
The learning curve might be steeper than with other software, but with R, the results of your analysis do not rely on remembering a succession of pointing and clicking, but instead on a series of written commands, and that’s a good thing! Ainsi, si vous souhaitez refaire votre analyse parce que vous avez collecté plus de données, vous n’avez pas besoin de vous rappeler sur quel bouton vous avez cliqué dans quel ordre pour obtenir vos résultats ; il vous suffit de réexécuter votre script.
Travailler avec des scripts rend les étapes que vous avez utilisées dans votre analyse claires, et le code que vous écrivez peut être inspecté par quelqu’un d’autre qui peut vous donner des commentaires et repérer les erreurs.
Travailler avec des scripts vous oblige à avoir une compréhension plus profonde de ce que vous faites et facilite votre apprentissage et votre compréhension des méthodes que vous utilisez.
Le code R est idéal pour la reproductibilité
La reproductibilité signifie que quelqu’un d’autre (y compris votre futur moi) peut obtenir les mêmes résultats à partir du même ensemble de données en utilisant le même code d’analyse .
R s’intègre à d’autres outils pour générer des manuscrits ou des rapports à partir de votre code . Si vous collectez plus de données ou corrigez une erreur dans votre ensemble de données, les chiffres et les tests statistiques de votre manuscrit ou rapport sont mis à jour automatiquement.
Un nombre croissant de revues et d’agences de financement s’attendent à ce que les analyses soient reproductibles, donc connaître R vous donnera un avantage avec ces exigences.
R est interdisciplinaire et extensible
Avec plus de 10 000 packages2 pouvant être installés pour étendre ses capacités, R fournit un cadre qui vous permet de combiner des approches statistiques de nombreuses disciplines scientifiques pour s’adapter au mieux à le cadre analytique dont vous avez besoin pour analyser vos données. Par exemple, R propose des packages pour l’analyse d’images, le SIG, les séries chronologiques, la génétique de population et bien plus encore.
, the Comprehensive R Archive Network. From the R Journal, Volume 10/2, December 2018.](../fig/cran.png)
R fonctionne sur des données de toutes formes et tailles
Les compétences que vous apprenez avec R évoluent facilement avec la taille de votre ensemble de données . Que votre ensemble de données comporte des centaines ou des millions de lignes, cela ne fera pas beaucoup de différence pour vous.
R est conçu pour l’analyse des données. Il est livré avec des structures de données spéciales et des types de données qui facilitent la gestion des données manquantes et des facteurs statistiques .
R peut se connecter à des feuilles de calcul, des bases de données et à de nombreux autres formats de données, sur votre ordinateur ou sur le Web.
R produit des graphiques de haute qualité
Les fonctionnalités de traçage de R sont étendues et vous permettent d’ajuster n’importe quel aspect de votre graphique pour transmettre le plus efficacement possible le message de vos données.
R a une communauté nombreuse et accueillante
Des milliers de personnes utilisent R quotidiennement. Beaucoup d’entre eux sont prêts à vous aider via des listes de diffusion et des sites Web tels que Stack Overflow, ou sur le RStudio communauté. Ces larges communautés d’utilisateurs s’étendent à des domaines spécialisés tels que la bioinformatique. L’un de ces sous-ensembles de la communauté R est Bioconductor, un projet scientifique pour l’analyse et la compréhension « des données provenant d’essais biologiques actuels et émergents ». Cet atelier a été développé par des membres de la communauté Bioconductor ; pour plus d’informations sur Bioconductor, veuillez consulter l’atelier complémentaire “The Bioconductor Project”.
Non seulement R est gratuit, mais il est également open source et multiplateforme
N’importe qui peut inspecter le code source pour voir comment R fonctionne. Grâce à cette transparence, il y a moins de risques d’erreurs, et si vous (ou quelqu’un d’autre) en trouvez, vous pouvez signaler et corriger des bugs.
Connaître RStudio
Commençons par découvrir RStudio, qui est un environnement de développement intégré (IDE) permettant de travailler avec R.
Le produit open source RStudio IDE est gratuit sous la Affero General Public License (AGPL) v3. L’IDE RStudio est également disponible avec une licence commerciale et une assistance prioritaire par courrier électronique de Posit, Inc.
Nous utiliserons l’IDE RStudio pour écrire du code, parcourir les fichiers sur notre ordinateur, inspecter les variables que nous allons créer et visualiser les tracés que nous allons générer. RStudio peut également être utilisé pour d’autres choses (par exemple, le contrôle de version, le développement de packages, l’écriture d’applications Shiny) que nous n’aborderons pas pendant l’atelier.
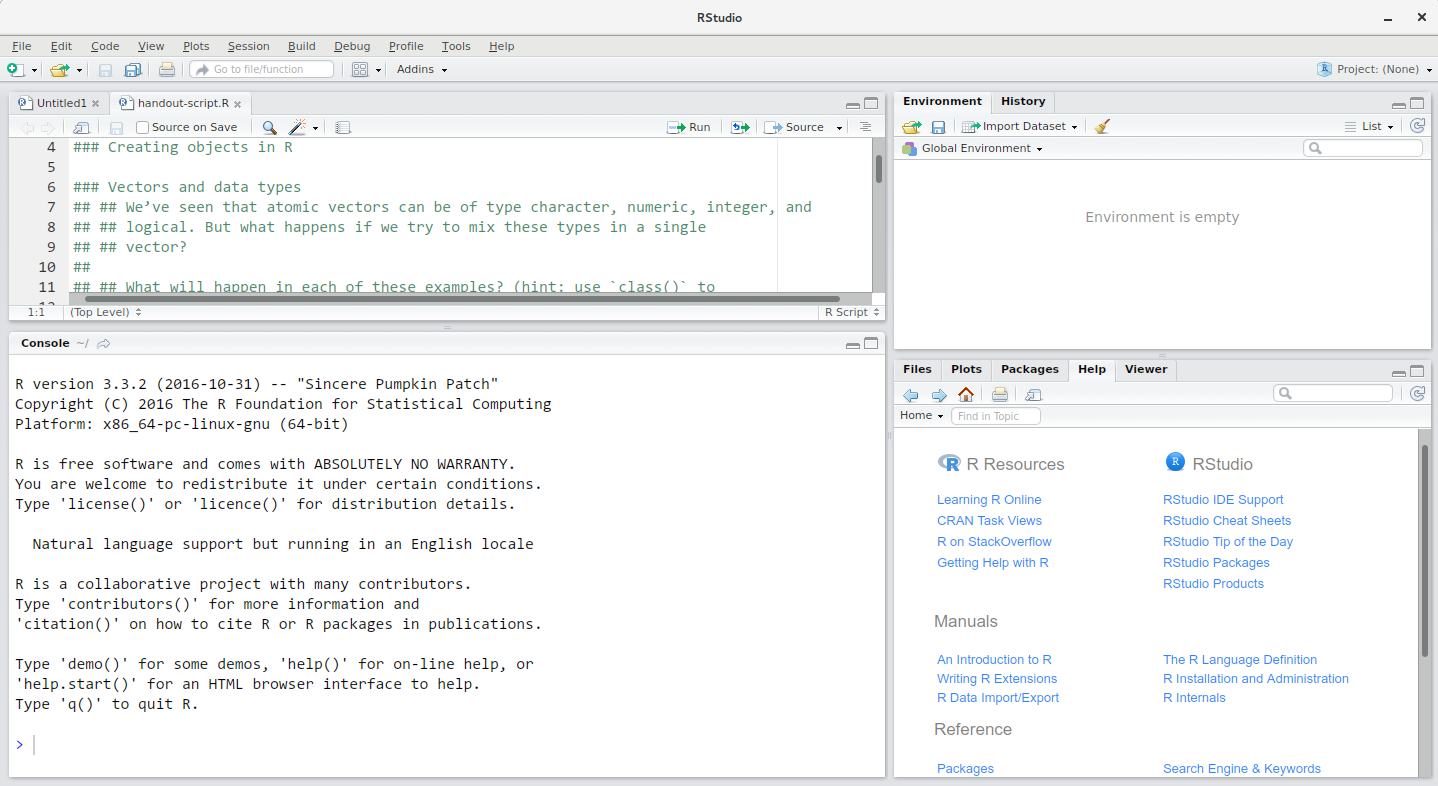
La fenêtre RStudio est divisée en 4 “Volets” :
- la Source de vos scripts et documents (en haut à gauche, dans la mise en page par défaut )
- votre Environnement/Historique (en haut à droite),
- vos Fichiers/Tracés/Packages/Aide/Visionneuse (en bas à droite), et
- la R Console (en bas à gauche).
L’emplacement de ces volets et leur contenu peuvent être
personnalisés (voir le menu ,
Outils -> Options globales -> Disposition des volets).
L’un des avantages de l’utilisation de RStudio est que toutes les informations dont vous avez besoin pour écrire du code sont disponibles dans une seule fenêtre. De plus, avec de nombreux raccourcis, la complétion automatique et la mise en surbrillance pour les principaux types de fichiers que vous utilisez lors du développement dans R, RStudio facilitera la saisie de et moins sujet aux erreurs.
Mise en place
Il est recommandé de conserver un ensemble de données, d’analyses et de textes connexes autonomes dans un seul dossier, appelé **répertoire de travail **. Tous les scripts de ce dossier peuvent alors utiliser chemins relatifs vers les fichiers qui indiquent où dans le projet se trouve un fichier (par opposition aux chemins absolus, qui pointent vers l’endroit où se trouve un fichier ). se trouve sur un ordinateur spécifique). Travailler de cette façon rend beaucoup plus facile le déplacement de votre projet sur votre ordinateur et le partage avec d’autres sans vous soucier de savoir si les scripts sous-jacents fonctionneront toujours.
RStudio fournit un ensemble d’outils utiles pour ce faire via son interface “Projets” , qui non seulement crée un répertoire de travail pour vous, mais mémorise également son emplacement (vous permettant d’y accéder rapidement ) et conserve éventuellement les paramètres personnalisés et les fichiers ouverts pour faciliter la reprise du travail après une pause. Suivez les étapes de création d’un “Projet R” pour ce tutoriel ci-dessous.
- Démarrez RStudio.
- Dans le menu « Fichier », cliquez sur « Nouveau projet ». Choisissez
Nouveau répertoire, puisNouveau projet. - Entrez un nom pour ce nouveau dossier (ou “répertoire”) et
choisissez un emplacement pratique pour celui-ci. Ce sera votre
répertoire de travail pour cette session (ou tout le
cours) (par exemple,
bioc-intro). - Cliquez sur « Créer un projet ».
- (Facultatif) Définissez les préférences sur « Jamais » pour enregistrer l’espace de travail dans RStudio.
Les préférences par défaut de RStudio fonctionnent généralement bien, mais enregistrer un espace de travail dans .RData peut être fastidieux, surtout si vous travaillez avec des ensembles de données plus volumineux. Pour désactiver cela, allez dans Outils –> « Options globales » et sélectionnez l’option « Jamais » pour « Enregistrer l’espace de travail dans .RData » à la sortie.

Pour éviter les problèmes d’encodage des caractères entre Windows et d’autres systèmes d’exploitation, nous allons définir UTF-8 par défaut :
Organiser votre répertoire de travail
L’utilisation d’une structure de dossiers cohérente dans vos projets aidera à garder les choses organisées et facilitera également la recherche/le classement des éléments à l’avenir. Ce peut être particulièrement utile lorsque vous avez plusieurs projets. En général, vous pouvez créer des répertoires (dossiers) pour les scripts, données et documents.
-
data/Utilisez ce dossier pour stocker vos données brutes et les ensembles de données intermédiaires que vous pouvez créer pour les besoins d’une analyse particulière. Par par souci de transparence et de provenance, vous devez toujours conserver une copie de votre données brutes accessibles et effectuez autant de le nettoyage et le prétraitement de vos données par programme (c’est-à-dire avec scripts, plutôt que manuellement) que possible. Séparer les données brutes des données traitées est également une bonne idée. Par exemple, vous pourriez avoir les fichiersdata/raw/tree_survey.plot1.txtet...plot2.txtconservés séparés d’undata/processed/tree.survey. fichier csvgénéré par le scriptscripts/01.preprocess.tree_survey.R. -
documents/Ce serait un endroit pour conserver les plans, les brouillons, les et d’autres textes. -
scripts/(ousrc) Ce serait l’emplacement où conserver vos scripts R pour différentes analyses ou traçages, et potentiellement un dossier séparé pour vos fonctions (plus nous y reviendrons plus tard).
Vous souhaiterez peut-être des répertoires ou sous-répertoires supplémentaires en fonction de les besoins de votre projet, mais ceux-ci devraient constituer l’épine dorsale de votre répertoire de travail .
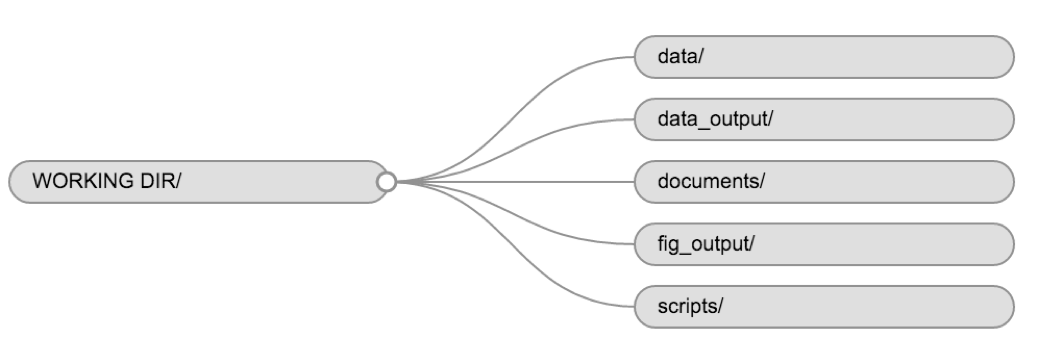
Pour ce cours, nous aurons besoin d’un dossier data/
pour stocker nos données brutes, et nous utiliserons
data_output/ lorsque nous apprendrons à exporter des
données sous forme de fichiers CSV, et Dossier fig_output/
pour les figures que nous allons enregistrer.
Défi : créer la structure de répertoires de votre projet
Sous l’onglet « Fichiers » à droite de l’écran, cliquez sur « Nouveau
dossier » et créez un dossier nommé « données » dans votre répertoire de
travail nouvellement créé (par exemple, « ~/bioc
-intro/données). (Vous pouvez également taperdir.create(“data”)sur votre console R.) Répétez ces opérations pour créer un dossierdata_output/et unfig_output`.
Nous allons conserver le script à la racine de notre répertoire de travail car nous n’allons utiliser qu’un seul fichier et cela rendra les choses plus faciles.
Votre répertoire de travail devrait maintenant ressembler à ceci :
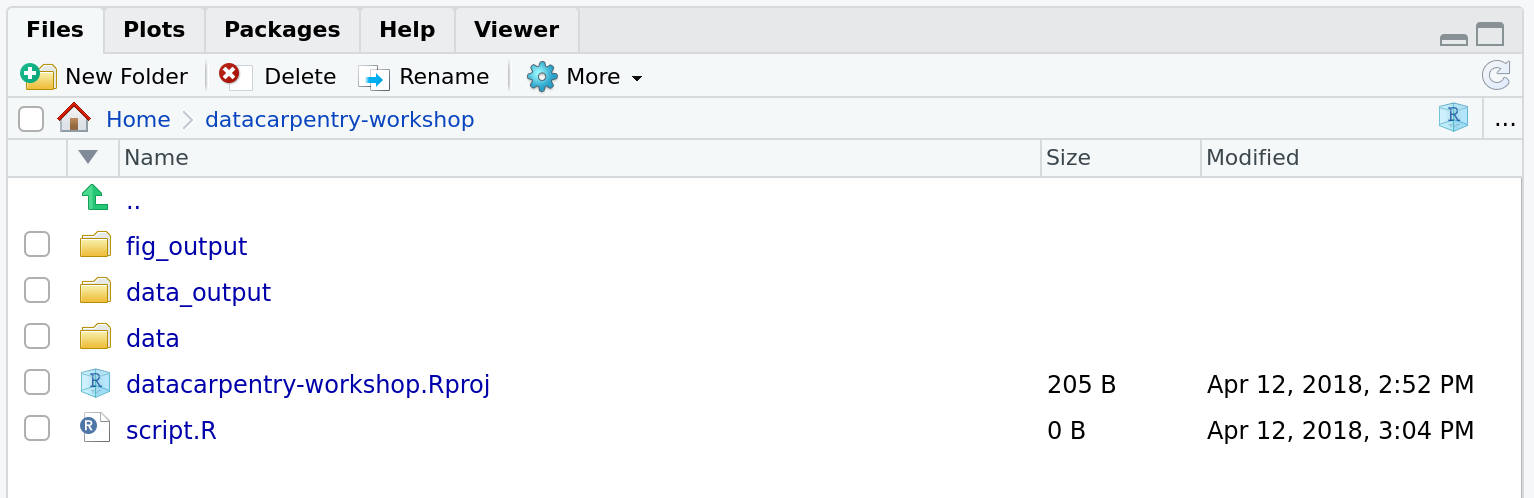
La gestion de projet s’applique également aux projets de bioinformatique, bien sûr3. William Noble (@Noble:2009) propose la structure de répertoires suivante :
Les noms de répertoires sont en gros caractères et les noms de fichiers sont en caractères plus petits . Seul un sous-ensemble des fichiers est affiché ici. Notez que les dates sont formatées
<year>-<month>-<day>afin qu’elles puissent être triées par ordre chronologique. Le code sourcesrc/ms-analysis.cest compilé pour créerbin/ms-analysiset est documenté dansdoc/ms-analysis.html. Les fichiersREADMEdans les répertoires de données précisent qui a téléchargé les fichiers de données à partir de quelle URL et à quelle date . Le script du piloteresults/2009-01-15/runallgénère automatiquement les trois sous-répertoires split1, split2 et split3, correspondant à trois divisions de validation croisée. Le scriptbin/parse-sqt.pyest appelé par les deux scripts du piloterunall.
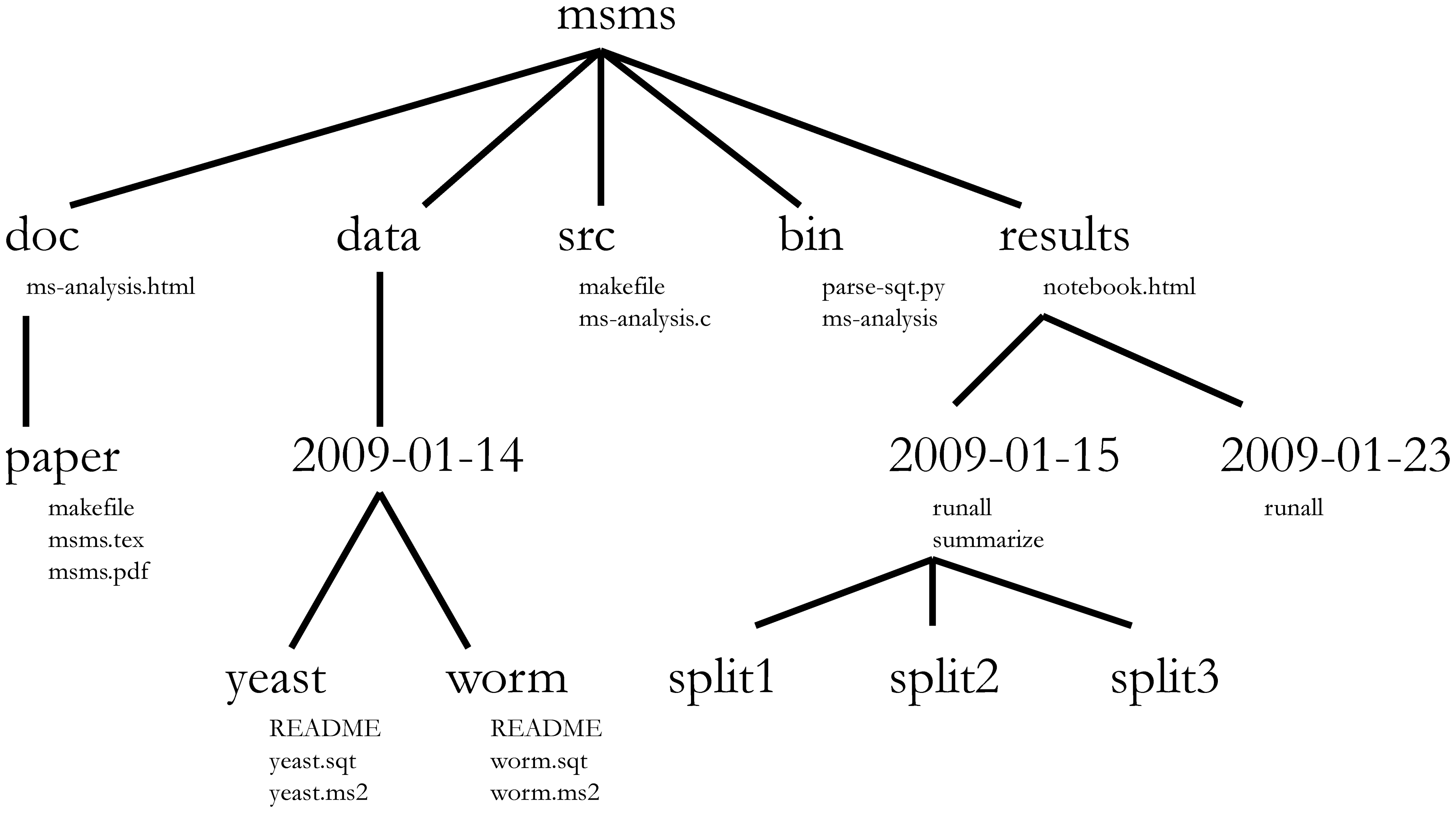
L’aspect le plus important d’un répertoire de projet bien défini et bien documenté est de permettre à quelqu’un qui n’est pas familier avec le projet 4 de
comprendre en quoi consiste le projet, quelles données sont disponibles, quelles analyses ont été effectuées et quels résultats ont été produits et, plus important encore,
répétez l’analyse à nouveau - avec de nouvelles données ou en modifiant certains paramètres d’analyse .
Le répertoire de travail
Le répertoire de travail est un concept important à comprendre. C’est l’endroit à partir duquel R recherchera et enregistrera les fichiers. Lorsque vous écrivez du code pour votre projet, il doit faire référence à des fichiers en relation avec la racine de votre répertoire de travail et n’a besoin que de fichiers au sein de cette structure .
L’utilisation de projets RStudio facilite cela et garantit que votre
répertoire de travail est correctement défini. Si vous avez besoin de le
vérifier, vous pouvez utiliser getwd(). If for some reason
your working directory is not what it should be, you can change it in
the RStudio interface by navigating in the file browser where your
working directory should be, and clicking on the blue gear icon
More, and select Set As Working Directory.
Vous pouvez également utiliser
setwd("/path/to/working/directory") pour réinitialiser
votre répertoire de travail. Cependant, vos scripts ne doivent pas
inclure cette ligne car elle échouera sur l’ordinateur de quelqu’un
d’autre.
Exemple
Le schéma ci-dessous représente le répertoire de travail
bioc-intro avec les sous-répertoires data et
fig_output, et 2 fichiers dans ce dernier :
bioc-intro/data/
/fig_output/fig1.pdf
/fig_output/fig2.pngSi on était dans le répertoire de travail, on pourrait faire
référence au fichier fig1.pdf en utilisant le chemin
relatif bioc-intro/fig_output/fig1.pdf ou le chemin absolu
/ accueil/user/bioc-intro/fig_output/fig1.pdf.
Si nous étions dans le répertoire data, nous
utiliserions le chemin relatif ../fig_output/fig1.pdf ou le
même chemin absolu
/home/user/bioc-intro /fig_output/fig1.pdf.
Interagir avec R
La base de la programmation est que nous écrivons les instructions que l’ordinateur doit suivre, puis nous disons à l’ordinateur de suivre ces instructions . Nous écrivons, ou codeons, des instructions dans R car c’est un langage commun que l’ordinateur et nous pouvons comprendre. Nous appelons les instructions commandes et nous disons à l’ordinateur de suivre les instructions en exécutant (également appelé exécutant) ces commandes.
Il existe deux manières principales d’interagir avec R : en utilisant la console ou en utilisant des scripts (fichiers texte brut contenant votre code). Le volet de la console (dans RStudio, le panneau inférieur gauche) est l’endroit où les commandes écrites en langage R peuvent être saisies et exécutées immédiatement par l’ordinateur. C’est également là que les résultats seront affichés pour les commandes exécutées. Vous pouvez taper des commandes directement dans la console et appuyer sur « Entrée » pour exécuter ces commandes , mais elles seront oubliées lorsque vous fermerez la session.
Parce que nous voulons que notre code et notre flux de travail soient reproductibles, il est préférable de taper les commandes souhaitées dans l’éditeur de script et d’enregistrer le script . De cette façon, il existe un enregistrement complet de ce que nous avons fait, et n’importe qui (y compris notre futur moi !) peuvent facilement reproduire les résultats sur leur ordinateur. Notez cependant que le simple fait de taper les commandes dans le script ne les exécute pas automatiquement - elles doivent quand même être envoyées à la console pour exécution.
RStudio vous permet d’exécuter des commandes directement depuis
l’éditeur de script en utilisant le raccourci Ctrl +
Entrée (sur Mac, Cmd + Return
fonctionnera également). La commande sur la ligne actuelle du script
(indiquée par le curseur) ou toutes les commandes dans le texte
actuellement sélectionné seront envoyées à la console et exécutées
lorsque vous appuyez sur Ctrl + Entrer. Vous
pouvez trouver d’autres raccourcis clavier dans cette aide-mémoire
RStudio sur l’IDE RStudio .
À un moment donné de votre analyse, vous souhaiterez peut-être
vérifier le contenu d’une variable ou la structure d’un objet, sans
nécessairement en conserver un enregistrement dans votre script. Vous
pouvez taper ces commandes et les exécuter directement dans la console.
RStudio fournit les raccourcis Ctrl + 1 et
Ctrl + 2 vous permettant de passer entre le
script et les volets de la console .
Si R est prêt à accepter les commandes, la console R affiche une
invite >. If it receives a command (by typing,
copy-pasting or sending from the script editor using Ctrl +
Enter), R will try to execute it, and when ready, will show
the results and come back with a new > prompt to wait
for new commands.
Si R attend toujours que vous saisissiez plus de données parce que
n’est pas encore terminé, la console affichera une invite « + ». Cela
signifie que vous n’avez pas fini de saisir une commande complète. This
is because you have not ‘closed’ a parenthesis or quotation, i.e. you
don’t have the same number of left-parentheses as right-parentheses, or
the same number of opening and closing quotation marks. Lorsque cela se
produit et que vous pensez avoir fini de taper votre commande, cliquez
dans la fenêtre de la console et appuyez sur « Échap » ; cela annulera
la commande incomplète et vous ramènera à l’invite
>.
Comment en savoir plus pendant et après le cours ?
Le matériel que nous aborderons au cours de ce cours vous donnera un premier aperçu de la façon dont vous pouvez utiliser R pour analyser des données pour votre propre recherche. Cependant, vous devrez en apprendre davantage pour effectuer des opérations avancées telles que nettoyer votre ensemble de données, utiliser des méthodes statistiques, ou créer de superbes graphiques[^dans ce cours]. La meilleure façon de devenir compétent et efficace en R, comme avec tout autre outil, est de l’utiliser pour répondre à vos questions de recherche réelles. En tant que débutant, il peut sembler intimidant de devoir écrire un script à partir de zéro, et étant donné que de nombreuses personnes rendent leur code disponible en ligne, modifiant le code existant pour répondre à vos objectifs. cela pourrait vous permettre de démarrer plus facilement.

Cherche de l’aide
Utilisez l’interface d’aide intégrée de RStudio pour rechercher plus d’informations sur les fonctions R.
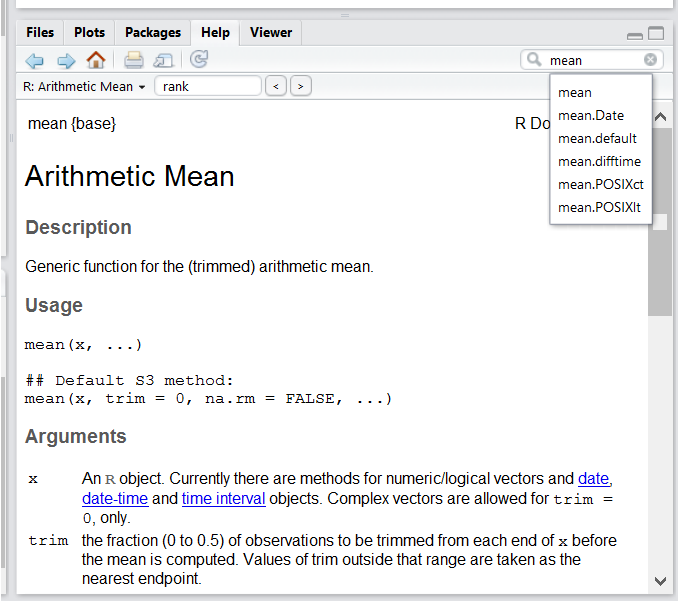
L’un des moyens les plus rapides d’obtenir de l’aide consiste à utiliser l’interface d’aide RStudio . Ce panneau par défaut se trouve dans le panneau inférieur droit de RStudio. Comme le montre la capture d’écran, en tapant le mot “Mean”, RStudio essaie également de donner un certain nombre de suggestions qui pourraient vous intéresser . La description s’affiche alors dans la fenêtre d’affichage .
Je connais le nom de la fonction que je souhaite utiliser, mais je ne sais pas comment l’utiliser
Si vous avez besoin d’aide avec une fonction spécifique, disons
barplot(), vous pouvez taper :
R
?barplot
Si vous avez juste besoin de vous rappeler les noms des arguments, vous pouvez utiliser :
R
args(lm)
Je veux utiliser une fonction qui fait X, il doit y avoir une fonction pour ça mais je ne sais pas laquelle…
Si vous recherchez une fonction pour effectuer une tâche
particulière, vous pouvez utiliser la fonction
help.search(), qui est appelée par le double point
d’interrogation ??. Cependant, cela ne recherche dans les
packages installés que les pages d’aide avec une correspondance avec
votre demande de recherche.
R
??kruskal
Si vous ne trouvez pas ce que vous cherchez, vous pouvez utiliser le site Web rdocumentation.org qui recherche dans les fichiers d’aide de tous les forfaits disponibles.
Enfin, une recherche générique sur Google ou sur Internet “R <task>” vous enverra souvent soit à la documentation du package appropriée, soit à un forum utile où quelqu’un d’autre a déjà posé votre question.
Je suis coincé… Je reçois un message d’erreur que je ne comprends pas
Commencez par rechercher le message d’erreur sur Google. Cependant, cela ne fonctionne pas toujours très bien car souvent, les développeurs de packages s’appuient sur la détection d’erreurs fournie par R. Vous vous retrouvez avec des messages d’erreur généraux qui pourraient ne pas être très utiles pour diagnostiquer un problème. problème (par exemple “indice hors limites”). Si le message est très générique, vous pouvez également inclure le nom de la fonction ou du package que vous utilisez dans votre requête.
Cependant, vous devriez vérifier Stack Overflow. Recherchez en
utilisant la balise [r]. La plupart des questions ont déjà
reçu une réponse, mais le défi consiste à utiliser les bons mots dans la
recherche pour trouver les réponses :
http://stackoverflow.com/questions/tagged/r
The Introduction to R can also be dense for people with little programming experience but it is a good place to understand the underpinnings of the R language.
La FAQ R est dense et technique mais elle regorge d’informations utiles.
Demander de l’aide
La clé pour recevoir de l’aide de quelqu’un est qu’il comprenne rapidement votre problème. Vous devez faire en sorte qu’il soit aussi simple que possible d’identifier où pourrait se situer le problème.
Essayez d’utiliser les mots corrects pour décrire votre problème. Par exemple, un package n’est pas la même chose qu’une bibliothèque. La plupart des gens comprendront ce que vous vouliez dire, mais d’autres ont des sentiments très forts à propos de la différence de sens. Le point clé est que cela peut rendre les choses déroutantes pour les personnes qui essaient de vous aider. Soyez aussi précis que possible lorsque vous décrivez votre problème.
Si possible, essayez de réduire ce qui ne fonctionne pas à un simple *exemple reproductible *. Si vous pouvez reproduire le problème en utilisant un très petit cadre de données au lieu de celui de 50 000 lignes et 10 000 colonnes, fournissez le petit avec la description de votre problème. Le cas échéant, essayez de généraliser ce que vous faites afin que même les personnes qui ne font pas partie de votre domaine puissent comprendre la question. Par exemple, au lieu d’utiliser un sous-ensemble de votre ensemble de données réel, créez un petit (3 colonnes, 5 lignes) générique. Pour plus d’informations sur la façon d’écrire un exemple reproductible, voir cet article de Hadley Wickham.
Pour partager un objet avec quelqu’un d’autre, s’il est relativement
petit, vous pouvez utiliser la fonction dput(). Il produira
du code R qui peut être utilisé pour recréer exactement le même objet
que celui en mémoire :
R
## iris is an example data frame that comes with R and head() is a
## function that returns the first part of the data frame
dput(head(iris))
SORTIE
structure(list(Sepal.Length = c(5.1, 4.9, 4.7, 4.6, 5, 5.4),
Sepal.Width = c(3.5, 3, 3.2, 3.1, 3.6, 3.9), Petal.Length = c(1.4,
1.4, 1.3, 1.5, 1.4, 1.7), Petal.Width = c(0.2, 0.2, 0.2,
0.2, 0.2, 0.4), Species = structure(c(1L, 1L, 1L, 1L, 1L,
1L), levels = c("setosa", "versicolor", "virginica"), class = "factor")), row.names = c(NA,
6L), class = "data.frame")If the object is larger, provide either the raw file (i.e., your CSV file) with your script up to the point of the error (and after removing everything that is not relevant to your issue). Alternativement, en particulier si votre question n’est pas liée à un bloc de données, vous pouvez enregistrer n’importe quel objet R dans un fichier[^export] :
R
saveRDS(iris, file="/tmp/iris.rds")
Le contenu de ce fichier n’est cependant pas lisible par l’homme et
ne peut pas être publié directement sur Stack Overflow. Au lieu de cela,
il peut être envoyé à quelqu’un par email qui pourra le lire avec la
commande readRDS() (ici, suppose que le fichier téléchargé
se trouve dans un dossier Téléchargements dans le
répertoire personnel de l’utilisateur) :
R
some_data <- readRDS(file="~/Downloads/iris.rds")
Dernier point, mais non le moindre, incluez toujours la
sortie de sessionInfo() car elle fournit des
informations critiques sur votre plate-forme, les versions de R et les
packages que vous utilisez. utilisation, et d’autres informations qui
peuvent être très utiles pour comprendre votre problème.
R
sessionInfo()
SORTIE
R version 4.4.1 (2024-06-14)
Platform: x86_64-pc-linux-gnu
Running under: Ubuntu 22.04.4 LTS
Matrix products: default
BLAS: /usr/lib/x86_64-linux-gnu/blas/libblas.so.3.10.0
LAPACK: /usr/lib/x86_64-linux-gnu/lapack/liblapack.so.3.10.0
locale:
[1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
[4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
[7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
[10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
time zone: Asia/Tokyo
tzcode source: system (glibc)
attached base packages:
[1] stats graphics grDevices utils datasets methods base
loaded via a namespace (and not attached):
[1] compiler_4.4.1 tools_4.4.1 highr_0.11 knitr_1.48
[5] xfun_0.47 evaluate_0.24.0Où demander de l’aide ?
- La personne assise à côté de vous pendant le cours. N’hésitez pas à parler à votre voisin pendant l’atelier, comparer vos réponses, et demander de l’aide.
- Vos collègues amicaux : si vous connaissez quelqu’un avec plus d’expérience que vous, il pourra et voudra peut-être vous aider.
- Stack Overflow : si votre question n’a pas reçu de réponse auparavant et est bien conçue, il y a de fortes chances que vous obteniez un réponse en moins de 5 minutes. N’oubliez pas de suivre leurs directives sur comment poser une bonne question.
- La liste de diffusion R-help : elle est lue par un grand nombre de personnes (dont la plupart des l’équipe principale de R), beaucoup de gens y publient des messages, mais le ton peut être assez sec, et il n’est pas toujours très accueillant pour les nouveaux utilisateurs. Si votre question est valide, vous avez de chances d’obtenir une réponse très rapidement, mais ne vous attendez pas à ce qu’elle vienne avec des visages souriants. Aussi, ici plus qu’ailleurs, veillez à d’utiliser un vocabulaire correct (sinon vous pourriez obtenir une réponse pointant vers une mauvaise utilisation de vos mots plutôt que de répondre à votre question). Vous aurez également plus de succès si votre question concerne une fonction de base plutôt qu’un package spécifique.
- Si votre question concerne un package spécifique, vérifiez s’il
existe une liste de diffusion pour celui-ci. Habituellement, il est
inclus dans le fichier DESCRIPTION du package accessible en utilisant
packageDescription("name-of-package"). Vous pouvez également essayer d’envoyer un e-mail directement à l’auteur du package ou d’ouvrir un ticket sur le référentiel de code (par exemple, GitHub). - Il existe également quelques listes de diffusion thématiques (SIG, phylogénétique, etc…), la liste complète est ici.
Davantage de ressources
Le Guide de publication pour les listes de diffusion R.
-
Comment demander de l’aide R
directives utiles.
Ce billet de blog de Jon Skeet contient des conseils assez complets sur la façon dont pour poser des questions de programmation.
Le package reprex est très utile pour créer des exemples reproductibles lorsque vous demandez de l’aide à . The rOpenSci community call “How to ask questions so they get answered” (Github link and video recording) includes a presentation of the reprex package and of its philosophy.
Forfaits R
Chargement des paquets
Comme nous l’avons vu plus haut, les packages R jouent un rôle
fondamental dans R. Les utilisent les fonctionnalités d’un package, en
supposant qu’il soit installé, il faut d’abord le charger pour pouvoir
l’utiliser . Cela se fait avec la fonction library().
Ci-dessous, nous chargeons ggplot2.
R
library("ggplot2")
Installation des packages
Le référentiel de packages par défaut est The Comprehensive R
Archive Network (CRAN), et tout package disponible sur CRAN peut
être installé avec la fonction install.packages().
Ci-dessous, par exemple, , nous installons le package dplyr
que nous découvrirons plus tard.
R
install.packages("dplyr")
Cette commande installera le package dplyr ainsi que
toutes ses dépendances, c’est à dire tous les packages sur lesquels il
s’appuie pour fonctionner.
Un autre référentiel majeur de packages R est géré par Bioconductor.
Packages
Bioconductor sont gérés et installés à l’aide d’un package dédié, à
savoir BiocManager, qui peut être installé à partir de CRAN
avec
R
install.packages("BiocManager")
Des packages individuels tels que SummarizedExperiment
(nous l’utiliserons plus tard), DESeq2 (pour l’analyse
RNA-Seq) et tout autre de Bioconductor ou CRAN peuvent ensuite être
installés avec BiocManager :: installer.
R
BiocManager::install("SummarizedExperiment")
BiocManager::install("DESeq2")
Par défaut, BiocManager::install() vérifiera également
tous vos packages installés et verra si des versions plus récentes sont
disponibles. S’il y en a, il vous les montrera et vous demandera si vous
souhaitez « Mettre à jour tout/certains/aucun ? [a/s/n] :` et attendez
votre réponse. Bien que vous deviez vous efforcer de disposer des
versions de packages les plus à jour, en pratique, nous vous
recommandons de mettre à jour les packages uniquement lors d’une
nouvelle session R avant le chargement des packages.
Au lieu d’utiliser R directement depuis la console de ligne de commande . Il existe d’autres logiciels qui s’interfacent et intègrent avec R, mais RStudio est particulièrement bien adapté aux débutants tout en proposant de nombreuses fonctionnalités très avancées.↩︎
c’est-à-dire des modules complémentaires qui confèrent à R de nouvelles fonctionnalités, telles que l’analyse de données bioinformatiques.↩︎
Dans ce cours, nous considérons la bioinformatique comme une science des données appliquée aux données biologiques ou bio-médicales.↩︎
Cette personne pourrait être, et sera très probablement votre futur moi, quelques mois ou années après que les analyses aient été effectuées.↩︎
Content from Introduction à R
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 120 minutes
Vue d'ensemble
Questions
- Premières commandes dans R
Objectifs
- Définissez les termes suivants relatifs à R : objet, affectation, appel, fonction, arguments, options.
- Attribuez des valeurs aux objets dans R.
- Apprenez à nommer des objets
- Utilisez les commentaires pour informer le script.
- Résoudre des opérations arithmétiques simples dans R.
- Appelez des fonctions et utilisez des arguments pour modifier leurs options par défaut.
- Inspectez le contenu des vecteurs et manipulez leur contenu.
- Sous-ensembler et extraire des valeurs à partir de vecteurs.
- Analysez les vecteurs avec des données manquantes.
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Créer des objets dans R
Vous pouvez obtenir le résultat de R simplement en tapant math dans la console :
R
3 + 5
SORTIE
[1] 8R
12 / 7
SORTIE
[1] 1.714286Cependant, pour faire des choses utiles et intéressantes, nous devons
attribuer des valeurs à objets. Pour créer un objet,
nous devons lui donner un nom suivi de l’opérateur d’affectation
<-, et de la valeur que nous voulons lui donner :
R
poids_kg <- 55
<- est l’opérateur d’affectation. Il attribue des
valeurs à droite aux objets à gauche. Ainsi, après avoir exécuté
x <- 3, la valeur de x est 3.
La flèche peut être lue comme 3 entre dans
x. Pour des raisons historiques , vous pouvez également
utiliser = pour les affectations, mais pas dans tous les
contextes . En raison du légères
différences dans la syntaxe, il est une bonne pratique de toujours
utiliser <- pour les affectations.
In RStudio, typing Alt + - (push Alt
at the same time as the - key) will write <-
in a single keystroke in a PC, while typing Option +
- (push Option at the same time as the
- key) does the same in a Mac.
Nommer les variables
Les objets peuvent recevoir n’importe quel nom tel que « x », «
current_temperature » ou « subject_id ». Vous voulez que les noms de vos
objets soient explicites et pas trop longs. Ils ne peuvent pas commencer
par un nombre (2x n’est pas valide, mais x2
l’est). R est sensible à la casse (par exemple, weight_kg
est différent de Weight_kg). Certains noms ne peuvent pas
être utilisés car ils sont les noms de fonctions fondamentales dans R
(par exemple, if, else, for, voir
ici
pour une liste complète). En général, même si c’est autorisé, il est
préférable de de ne pas utiliser d’autres noms de fonctions (par
exemple, c, T, mean,
data, df, poids). En cas de
doute, consultez l’aide pour voir si le nom est déjà utilisé. Il est
également préférable d’éviter les points (.) dans un nom
d’objet comme dans my.dataset. There are many functions in
R with dots in their names for historical reasons, but because dots have
a special meaning in R (for methods) and other programming languages,
it’s best to avoid them. Il est également recommandé d’utiliser des noms
pour les noms d’objets et des verbes pour les noms de fonctions. Il est
important d’être cohérent dans le style de votre code (où vous placez
les espaces, comment vous nommez les objets, etc.). L’utilisation d’un
style de codage cohérent rend votre code plus clair à lire pour votre
futur moi et vos collaborateurs. Dans R, certains guides de style
populaires sont de Google, le
tidyverse et le
Bioconductor style guide. Le Tidyverse est très complet et peut sembler
écrasant au début . Vous pouvez installer le package lintr
pour vérifier automatiquement les problèmes dans le style de votre
code.
Objets et variables : ce que l’on appelle des « objets » dans « R » sont connus sous le nom de « variables » dans de nombreux autres langages de programmation. Selon le contexte, « objet » et « variable » peuvent avoir des significations radicalement différentes. Cependant, dans cette leçon, les deux mots sont utilisés de manière synonyme. Pour plus d’informations voir ici.
Lors de l’attribution d’une valeur à un objet, R n’imprime rien. Vous pouvez forcer R à imprimer la valeur en utilisant des parenthèses ou en tapant le nom de l’objet :
R
weight_kg <- 55 # n'imprime rien
(weight_kg <- 55) # mais mettre des parenthèses autour de l'appel imprime la valeur de `weight_kg`
SORTIE
[1] 55R
weight_kg # et taper également le nom du objet
SORTIE
[1] 55Maintenant que R a « weight_kg » en mémoire, nous pouvons faire de l’arithmétique avec. Pour exemple, nous pouvons vouloir convertir ce poids en livres (le poids en livres est 2,2 fois le poids en kg) :
R
2.2 * weight_kg
SORTIE
[1] 121On peut également changer la valeur d’un objet en lui attribuant une nouvelle :
R
weight_kg <- 57.5
2.2 * weight_kg
SORTIE
[1] 126.5Cela signifie que l’attribution d’une valeur à un objet ne modifie
pas les valeurs de autres objets. Par exemple, stockons le poids de
l’animal en livres dans un nouvel objet , weight_lb :
R
weight_lb <- 2.2 * weight_kg
puis remplacez « weight_kg » par 100.
R
weight_kg <- 100
commentaires
Le caractère de commentaire dans R est #, tout ce qui se
trouve à droite d’un # dans un script sera ignoré par R. Il
est utile de laisser des notes et des explications dans vos scripts
.
RStudio permet de commenter ou décommenter facilement un paragraphe : après sélectionnant les lignes que vous souhaitez commenter, appuyez en même temps sur votre clavier Ctrl + Maj + C. If you only want to comment out one line, you can put the cursor at any location of that line (i.e. no need to select the whole line), then press Ctrl + Shift + C.
Fonctions et leurs arguments
Les fonctions sont des “scripts prédéfinis” qui automatisent des
ensembles de commandes plus complexes , y compris les affectations
d’opérations, etc. De nombreuses fonctions sont prédéfinies ou peuvent
être rendues disponibles en important des packages R (nous en
parlerons plus tard). Une fonction obtient généralement une ou plusieurs
entrées appelées arguments. Les fonctions renvoient souvent
(mais pas toujours) une valeur. Un exemple typique serait la
fonction sqrt(). L’entrée (l’argument) doit être un nombre
et la valeur de retour (en fait, la sortie ) est la racine carrée de ce
nombre. Exécuter une fonction (« l’exécuter ») est appelé
appeler la fonction. Un exemple d’appel de fonction est :
R
b <- sqrt(a)
Ici, la valeur de a est donnée à la fonction
sqrt(), la fonction sqrt() calcule la racine
carrée, et renvoie la valeur qui est ensuite attribuée à l’objet ‘b’.
Cette fonction est très simple car elle ne prend qu’un seul
argument.
The return ‘value’ of a function need not be numerical (like that of
sqrt()), and it also does not need to be a single item: it
can be a set of things, or even a dataset. Nous le verrons lorsque nous
lirons des fichiers de données dans R.
Les arguments peuvent être n’importe quoi, non seulement des nombres ou des noms de fichiers, mais aussi d’autres objets . La signification exacte de chaque argument diffère selon la fonction et doit être recherchée dans la documentation (voir ci-dessous). Certaines fonctions prennent des arguments qui peuvent soit être spécifiés par l’utilisateur, soit, s’ils sont laissés de côté, prendre une valeur par défaut : ceux-ci sont appelés options. Les options sont généralement utilisées pour modifier le fonctionnement de la fonction , par exemple si elle ignore les « mauvaises valeurs » ou quel symbole utiliser dans un tracé. Cependant, si vous souhaitez quelque chose de spécifique, vous pouvez spécifier une valeur de votre choix qui sera utilisée à la place de la valeur par défaut.
Essayons une fonction qui peut prendre plusieurs arguments :
round().
R
round(3.14159)
SORTIE
[1] 3Ici, nous avons appelé round() avec un seul argument,
3.14159, et il a renvoyé la valeur 3. En
effet, la valeur par défaut est d’arrondir au nombre entier le plus
proche. Si nous voulons plus de chiffres, nous pouvons voir comment
procéder en obtenant des informations sur la fonction
round. Nous pouvons utiliser args(round) ou
consulter l’aide pour cette fonction en utilisant
?round.
R
args(round)
SORTIE
function (x, digits = 0, ...)
NULLR
?round
Nous voyons que si nous voulons un nombre différent de chiffres, nous
pouvons taper digits=2 ou autant que nous le voulons.
R
round(3.14159, digits = 2)
SORTIE
[1] 3.14Si vous fournissez les arguments exactement dans le même ordre que celui dans lequel ils sont définis, vous n’avez pas besoin de les nommer :
R
round(3.14159, 2)
SORTIE
[1] 3.14Et si vous nommez les arguments, vous pouvez changer leur ordre :
R
round(digits = 2, x = 3.14159)
SORTIE
[1] 3.14Il est recommandé de placer les arguments non facultatifs (comme le nombre que vous arrondissez ) en premier dans votre appel de fonction et de spécifier les noms de tous les arguments facultatifs. Si vous ne le faites pas, quelqu’un qui lit votre code devra peut-être rechercher la définition d’une fonction avec des arguments inconnus pour comprendre ce que vous faites . En spécifiant le nom des arguments, vous protégez également contre d’éventuelles modifications futures dans l’interface de la fonction, qui peuvent potentiellement ajouter de nouveaux arguments entre ceux existants.
Vecteurs et types de données
Un vecteur est le type de données le plus courant et le plus basique
dans R, et est à peu près le cheval de bataille de R. Un vecteur est
composé d’une série de valeurs, telles que nombres ou caractères. Nous
pouvons attribuer une série de valeurs à un vecteur en utilisant la
fonction c(). Par exemple, nous pouvons créer un vecteur de
poids d’animaux et l’attribuer à un nouvel objet
weight_g :
R
weight_g <- c(50, 60, 65, 82)
weight_g
SORTIE
[1] 50 60 65 82Un vecteur peut également contenir des caractères :
R
molecules <- c("dna", "rna", "protein")
molecules
SORTIE
[1] "dna" "rna" "protein"Les guillemets autour de « adn », « arn », etc. sont ici essentiels. Sans les guillemets , R supposera qu’il existe des objets appelés « adn », « arn » et « protéine ». Comme ces objets n’existent pas dans la mémoire de R, il y aura un message d’erreur.
Il existe de nombreuses fonctions qui vous permettent d’inspecter le
contenu d’un vecteur . length() vous indique combien
d’éléments se trouvent dans un vecteur particulier :
R
length(weight_g)
SORTIE
[1] 4R
length(molecules)
SORTIE
[1] 3Une caractéristique importante d’un vecteur est que tous les éléments
sont du même type de données. La fonction class() indique
la classe (le type d’élément ) d’un objet :
R
class(weight_g)
SORTIE
[1] "numeric"R
class(molecules)
SORTIE
[1] "character"La fonction str() fournit un aperçu de la structure d’un
objet et de ses éléments. C’est une fonction utile lorsque vous
travaillez avec des objets volumineux et complexes :
R
str(weight_g)
SORTIE
num [1:4] 50 60 65 82R
str(molecules)
SORTIE
chr [1:3] "dna" "rna" "protein"Vous pouvez utiliser la fonction c() pour ajouter
d’autres éléments à votre vecteur :
R
weight_g <- c(weight_g, 90) # add to the end of the vector
weight_g <- c(30, weight_g) # add to the beginning of the vector
weight_g
SORTIE
[1] 30 50 60 65 82 90Dans la première ligne, nous prenons le vecteur d’origine
weight_g, ajoutons la valeur 90 à la fin de
celui-ci et enregistrons le résultat dans weight_g.
Ensuite, nous ajoutons la valeur « 30 » au début, en enregistrant à
nouveau le résultat dans « weight_g ».
Nous pouvons faire cela encore et encore pour développer un vecteur ou assembler un ensemble de données . Au fur et à mesure que nous programmons, cela peut être utile pour ajouter les résultats que nous collectons ou calculons.
Un vecteur atomique est le type de
données R le plus simple et est un vecteur linéaire d’un seul
type. Ci-dessus, nous avons vu 2 des 6 principaux types de vecteurs
**atomiques ** que R utilise : "caractère" et
"numérique" (ou "double"). Ce sont les
éléments de base à partir desquels tous les objets R sont construits.
Les 4 autres types de vecteurs atomiques sont :
-
"logique"pourTRUEetFALSE(le type de données booléen) -
"integer"pour les nombres entiers (par exemple,2L, leLindique à R que c’est un entier) -
"complexe"pour représenter des nombres complexes avec des parties réelles et imaginaires (par exemple,1 + 4i) et c’est tout ce que nous allons dire à leur sujet -
"raw"pour les bitstreams dont nous ne parlerons pas davantage
Vous pouvez vérifier le type de votre vecteur en utilisant la
fonction typeof() et en saisissant votre vecteur comme
argument.
Les vecteurs sont l’une des nombreuses structures de
données utilisées par R. Les autres importants sont les listes
(list), les matrices (matrix), les trames de
données (data.frame), les facteurs (factor) et
les tableaux (array ).
R les convertit implicitement pour qu’ils soient tous du même type
Défi:
Que se passera-t-il dans chacun de ces exemples ? (indice : utilisez
class() pour vérifier le type de données de vos objets et
tapez leurs noms pour voir ce qui se passe) :
R
num_char <- c(1, 2, 3, "a")
num_logical <- c(1, 2, 3, TRUE, FALSE)
char_logical <- c("a", "b", "c", TRUE)
tricky <- c(1, 2, 3, "4")
R
class(num_char)
SORTIE
[1] "character"R
num_char
SORTIE
[1] "1" "2" "3" "a"R
class(num_logical)
SORTIE
[1] "numeric"R
num_logical
SORTIE
[1] 1 2 3 1 0R
class(char_logical)
SORTIE
[1] "character"R
char_logical
SORTIE
[1] "a" "b" "c" "TRUE"R
class(tricky)
SORTIE
[1] "character"R
tricky
SORTIE
[1] "1" "2" "3" "4"Les vecteurs ne peuvent appartenir qu’à un seul type de données. R essaie de convertir (contraindre) le contenu de ce vecteur pour trouver un dénominateur commun qui ne perd aucune information.
Seulement un. Il n’y a pas de mémoire des types de données passés et
la coercition se produit la première fois que le vecteur est évalué. Par
conséquent, le TRUE dans num_logical est
converti en 1 avant d’être converti en "1"
dans combined_logical.
R
combined_logical
SORTIE
[1] "1" "2" "3" "1" "a" "b" "c" "TRUE"Défi:
Dans R, nous appelons la conversion d’objets d’une classe vers une autre classe coercition. Ces conversions se produisent selon une hiérarchie, selon laquelle certains types sont préférentiellement contraints vers d’autres types. Pouvez-vous dessiner un diagramme qui représente la hiérarchie de la façon dont ces types de données sont forcés ?
logique → numérique → caractère ← logique
Vecteurs de sous-ensemble
Si l’on veut extraire une ou plusieurs valeurs d’un vecteur, il faut fournir un ou plusieurs indices entre crochets. Par exemple:
R
molecules <- c("dna", "rna", "peptide", "protein")
molecules[2]
SORTIE
[1] "rna"R
molecules[c(3, 2)]
SORTIE
[1] "peptide" "rna" On peut également répéter les indices pour créer un objet avec plus d’éléments que celui d’origine :
R
more_molecules <- molecules[c(1, 2, 3, 2, 1, 4)]
more_molecules
SORTIE
[1] "dna" "rna" "peptide" "rna" "dna" "protein"Les indices R commencent à 1. Les langages de programmation comme Fortran, MATLAB, Julia et R commencent à compter à 1, car c’est ce que font généralement les êtres humains . Les langages de la famille C (y compris C++, Java, Perl, et Python) comptent à partir de 0 car c’est plus simple à faire pour les ordinateurs.
Enfin, il est également possible d’obtenir tous les éléments d’un vecteur sauf certains éléments spécifiés en utilisant des indices négatifs :
R
molecules ## all molecules
SORTIE
[1] "dna" "rna" "peptide" "protein"R
molecules[-1] ## all but the first one
SORTIE
[1] "rna" "peptide" "protein"R
molecules[-c(1, 3)] ## all but 1st/3rd ones
SORTIE
[1] "rna" "protein"R
molecules[c(-1, -3)] ## all but 1st/3rd ones
SORTIE
[1] "rna" "protein"Sous-ensemble conditionnel
Une autre méthode courante de sous-ensemble consiste à utiliser un
vecteur logique. TRUE sélectionnera l’élément avec le même
index, tandis que FALSE ne le fera pas :
R
weight_g <- c(21, 34, 39, 54, 55)
weight_g[c(TRUE, FALSE, TRUE, TRUE, FALSE)]
SORTIE
[1] 21 39 54Généralement, ces vecteurs logiques ne sont pas tapés à la main, mais sont la sortie d’autres fonctions ou tests logiques. Par exemple, si vous souhaitez sélectionner uniquement les valeurs supérieures à 50 :
R
## will return logicals with TRUE for the indices that meet
## the condition
weight_g > 50
SORTIE
[1] FALSE FALSE FALSE TRUE TRUER
## so we can use this to select only the values above 50
weight_g[weight_g > 50]
SORTIE
[1] 54 55Vous pouvez combiner plusieurs tests en utilisant &
(les deux conditions sont vraies, AND) ou | (au moins une
des conditions est vraie, OR) :
R
weight_g[weight_g < 30 | weight_g > 50]
SORTIE
[1] 21 54 55R
weight_g[weight_g >= 30 & weight_g == 21]
SORTIE
numeric(0)Ici, < signifie “inférieur à”, > pour
“supérieur à”, >= pour “supérieur ou égal à” et
== pour “égal à”. The double equal sign == is
a test for numerical equality between the left and right hand sides, and
should not be confused with the single = sign, which
performs variable assignment (similar to <-).
Une tâche courante consiste à rechercher certaines chaînes dans un
vecteur. On pourrait utiliser l’opérateur “ou” | pour
tester l’égalité de plusieurs valeurs, mais cela peut rapidement devenir
fastidieux. La fonction %in% permet de tester si l’un des
éléments d’un vecteur de recherche est trouvé :
R
molecules <- c("dna", "rna", "protein", "peptide")
molecules[molecules == "rna" | molecules == "dna"] # returns both rna and dna
SORTIE
[1] "dna" "rna"R
molecules %in% c("rna", "dna", "metabolite", "peptide", "glycerol")
SORTIE
[1] TRUE TRUE FALSE TRUER
molecules[molecules %in% c("rna", "dna", "metabolite", "peptide", "glycerol")]
SORTIE
[1] "dna" "rna" "peptide"R
"four" > "five"
SORTIE
[1] TRUELorsque vous utilisez > ou < sur des
chaînes, R compare leur ordre alphabétique. Ici, "quatre"
vient après "cinq", et est donc supérieur à .
Des noms
Il est possible de nommer chaque élément d’un vecteur. Le morceau de code ci-dessous montre un vecteur initial sans aucun nom, comment les noms sont définis et récupérés.
R
x <- c(1, 5, 3, 5, 10)
names(x) ## no names
SORTIE
NULLR
names(x) <- c("A", "B", "C", "D", "E")
names(x) ## now we have names
SORTIE
[1] "A" "B" "C" "D" "E"Lorsqu’un vecteur possède des noms, il est possible d’accéder aux éléments par leur nom , en plus de leur index.
R
x[c(1, 3)]
SORTIE
A C
1 3 R
x[c("A", "C")]
SORTIE
A C
1 3 Données manquantes
Comme R a été conçu pour analyser des ensembles de données, il inclut le concept de données manquantes (ce qui est rare dans d’autres langages de programmation ). Les données manquantes sont représentées dans les vecteurs par « NA ».
Lorsque vous effectuez des opérations sur des nombres, la plupart des
fonctions renverront « NA » si les données avec lesquelles vous
travaillez incluent des valeurs manquantes. Cette fonctionnalité rend
plus difficile l’ignorance des cas où vous avez affaire à données
manquantes. Vous pouvez ajouter l’argument na.rm = TRUE
pour calculer le résultat en ignorant les valeurs manquantes.
R
heights <- c(2, 4, 4, NA, 6)
mean(heights)
SORTIE
[1] NAR
max(heights)
SORTIE
[1] NAR
mean(heights, na.rm = TRUE)
SORTIE
[1] 4R
max(heights, na.rm = TRUE)
SORTIE
[1] 6Si vos données incluent des valeurs manquantes, vous souhaiterez
peut-être vous familiariser avec les fonctions is.na(),
na.omit() et complete.cases(). Voir ci-dessous
pour des exemples.
R
## Extract those elements which are not missing values.
heights[!is.na(heights)]
SORTIE
[1] 2 4 4 6R
## Returns the object with incomplete cases removed.
## The returned object is an atomic vector of type `"numeric"`
## (or `"double"`).
na.omit(heights)
SORTIE
[1] 2 4 4 6
attr(,"na.action")
[1] 4
attr(,"class")
[1] "omit"R
## Extract those elements which are complete cases.
## The returned object is an atomic vector of type `"numeric"`
## (or `"double"`).
heights[complete.cases(heights)]
SORTIE
[1] 2 4 4 6Défi:
- En utilisant ce vecteur de hauteurs en pouces, créez un nouveau vecteur en supprimant les NA.
R
heights <- c(63, 69, 60, 65, NA, 68, 61, 70, 61, 59, 64, 69, 63, 63, NA, 72, 65, 64, 70, 63, 65)
- Utilisez la fonction
median()pour calculer la médiane du vecteurheights. - Utilisez R pour déterminer combien de personnes dans l’ensemble mesurent plus de 67 pouces.
R
heights_no_na <- heights[!is.na(heights)]
## or
heights_no_na <- na.omit(heights)
R
median(heights, na.rm = TRUE)
SORTIE
[1] 64R
heights_above_67 <- heights_no_na[heights_no_na > 67]
length(heights_above_67)
SORTIE
[1] 6Génération de vecteurs
Constructeurs
Il existe quelques fonctions pour générer des vecteurs de différents
types. Pour générer un vecteur de valeurs numériques, on peut utiliser
le constructeur numeric() , fournissant la longueur du
vecteur de sortie comme paramètre . Les valeurs seront initialisées à
0.
R
numeric(3)
SORTIE
[1] 0 0 0R
numeric(10)
SORTIE
[1] 0 0 0 0 0 0 0 0 0 0Notez que si l’on demande un vecteur de numériques de longueur 0, on obtient exactement cela :
R
numeric(0)
SORTIE
numeric(0)Il existe des constructeurs similaires pour les caractères et les
logiques, nommés respectivement character() et
logical().
R
character(2) ## the empty character
SORTIE
[1] "" ""R
logical(2) ## FALSE
SORTIE
[1] FALSE FALSERépliquer des éléments
La fonction rep permet de répéter une valeur un certain
nombre de fois. Si nous voulons initier un vecteur de numériques de
longueur 5 avec la valeur -1, par exemple, nous pourrions faire ce qui
suit :
R
rep(-1, 5)
SORTIE
[1] -1 -1 -1 -1 -1De même, pour générer un vecteur rempli de valeurs manquantes, ce qui est souvent une bonne façon de commencer, sans poser d’hypothèses sur les données à collecter :
R
rep(NA, 5)
SORTIE
[1] NA NA NA NA NArep peut prendre en entrée des vecteurs de n’importe
quelle longueur (ci-dessus, nous avons utilisé des vecteurs de longueur
1) et de n’importe quel type. Par exemple, si nous voulons répéter cinq
fois les valeurs 1, 2 et 3, nous procéderions comme suit :
R
rep(c(1, 2, 3), 5)
SORTIE
[1] 1 2 3 1 2 3 1 2 3 1 2 3 1 2 3R
rep(c(1, 2, 3), each = 5)
SORTIE
[1] 1 1 1 1 1 2 2 2 2 2 3 3 3 3 3R
sort(rep(c(1, 2, 3), 5))
SORTIE
[1] 1 1 1 1 1 2 2 2 2 2 3 3 3 3 3Génération de séquence
Une autre fonction très utile est seq, pour générer une
séquence de nombres. Par exemple, pour générer une séquence d’entiers de
1 à 20 par pas de 2, on utiliserait :
R
seq(from = 1, to = 20, by = 2)
SORTIE
[1] 1 3 5 7 9 11 13 15 17 19La valeur par défaut de by est 1 et, étant donné que la
génération d’une séquence d’une valeur à une autre avec des pas de 1 est
fréquemment utilisée, il existe un raccourci :
R
seq(1, 5, 1)
SORTIE
[1] 1 2 3 4 5R
seq(1, 5) ## default by
SORTIE
[1] 1 2 3 4 5R
1:5
SORTIE
[1] 1 2 3 4 5Pour générer une séquence de nombres de 1 à 20 de longueur finale de 3, on utiliserait :
R
seq(from = 1, to = 20, length.out = 3)
SORTIE
[1] 1.0 10.5 20.0Échantillons aléatoires et permutations
Un dernier groupe de fonctions utiles sont celles qui génèrent des
données aléatoires . Le premier, sample, génère une
permutation aléatoire de un autre vecteur. Par exemple, pour tirer au
sort un ordre aléatoire de 10 étudiants à l’examen oral, j’attribue
d’abord à chaque étudiant un numéro de 1 à dix (par exemple en fonction
de l’ordre alphabétique de son nom) puis :
R
sample(1:10)
SORTIE
[1] 9 4 7 1 2 5 3 10 6 8Sans autres arguments, sample renverra une permutation
de tous les éléments du vecteur. Si je veux un échantillon aléatoire
d’une certaine taille, je définirais cette valeur comme deuxième
argument. Ci-dessous, j’échantillonne 5 lettres aléatoires de l’alphabet
contenu dans le vecteur letters prédéfini :
R
sample(letters, 5)
SORTIE
[1] "s" "a" "u" "x" "j"Si je voulais une sortie plus grande que le vecteur d’entrée, ou
pouvoir dessiner certains éléments plusieurs fois, je devrais définir
l’argument replace sur TRUE :
R
sample(1:5, 10, replace = TRUE)
SORTIE
[1] 2 1 5 5 1 1 5 5 2 2Défi:
En essayant les fonctions ci-dessus, vous aurez réalisé que les
échantillons sont effectivement aléatoires et qu’on n’obtient pas deux
fois la même permutation . Pour pouvoir reproduire ces tirages
aléatoires, on peut définir manuellement la graine de génération de
nombres aléatoires avec set.seed() avant de tirer
l’échantillon aléatoire.
Testez cette fonctionnalité avec votre voisin. Dessinez d’abord deux permutations aléatoires de « 1:10 » indépendamment et observez que vous obtenez résultats différents.
Définissez maintenant la graine avec, par exemple,
set.seed(123) et répétez le tirage au sort . Observez que
vous obtenez désormais les mêmes tirages au sort.
Répétez en définissant une graine différente.
Différentes permutations
R
sample(1:10)
SORTIE
[1] 9 1 4 3 6 2 5 8 10 7R
sample(1:10)
SORTIE
[1] 4 9 7 6 1 10 8 3 2 5Mêmes permutations avec la graine 123
R
set.seed(123)
sample(1:10)
SORTIE
[1] 3 10 2 8 6 9 1 7 5 4R
set.seed(123)
sample(1:10)
SORTIE
[1] 3 10 2 8 6 9 1 7 5 4Une graine différente
R
set.seed(1)
sample(1:10)
SORTIE
[1] 9 4 7 1 2 5 3 10 6 8R
set.seed(1)
sample(1:10)
SORTIE
[1] 9 4 7 1 2 5 3 10 6 8Extraire des échantillons à partir d’une distribution normale
La dernière fonction que nous allons voir est rnorm, qui
tire un échantillon aléatoire à partir d’une distribution normale. Deux
distributions normales de moyennes 0 et 100 et d’écarts types 1 et 5,
notées N(0, 1) et N(100, 5), sont présentées
ci-dessous.
Les trois arguments, n, mean et
sd, définissent la taille de l’échantillon , et les
paramètres de la distribution normale, c’est-à-dire la moyenne et son
écart type. Les valeurs par défaut de ce dernier sont 0 et 1.
R
rnorm(5)
SORTIE
[1] 0.69641761 0.05351568 -1.31028350 -2.12306606 -0.20807859R
rnorm(5, 2, 2)
SORTIE
[1] 1.3744268 -0.1164714 2.8344472 1.3690969 3.6510983R
rnorm(5, 100, 5)
SORTIE
[1] 106.45636 96.87448 95.62427 100.71678 107.12595Maintenant que nous avons appris à écrire des scripts et les bases des structures de données de R, nous sommes prêts à commencer à travailler avec des données plus volumineuses et à en apprendre davantage sur les trames de données.
Content from Commencer par les données
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 60 minutes
Vue d'ensemble
Questions
- Première analyse de données dans R
Objectifs
- Décrivez ce qu’est un « data.frame ».
- Chargez des données externes à partir d’un fichier .csv dans un bloc de données.
- Résumer le contenu d’un bloc de données.
- Décrivez ce qu’est un facteur.
- Convertissez entre les chaînes et les facteurs.
- Réorganisez et renommez les facteurs.
- Formater les dates.
- Exportez et enregistrez les données.
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Présentation des données d’expression des gènes
Nous allons utiliser une partie des données publiées par Blackmore , L’effet de l’infection des voies respiratoires supérieures sur les modifications transcriptomiques du SNC. Le but de l’étude était de déterminer l’effet d’une infection des voies respiratoires supérieures sur les modifications de la transcription de l’ARN se produisant dans le cervelet et la moelle épinière après l’infection. Gender matched eight week old C57BL/6 mice were inoculated with saline or with Influenza A by intranasal route and transcriptomic changes in the cerebellum and spinal cord tissues were evaluated by RNA-seq at days 0 (non-infected), 4 and 8.
L’ensemble de données est stocké sous forme de fichier CSV (valeurs séparées par des virgules). Chaque ligne contient des informations pour une seule mesure d’expression d’ARN, et les onze premières colonnes représentent :
| Colonne | Description |
|---|---|
| gène | Le nom du gène qui a été mesuré |
| échantillon | Le nom de l’échantillon dans lequel l’expression du gène a été mesurée |
| expression | La valeur de l’expression des gènes |
| organisme | L’organisme/l’espèce - ici toutes les données proviennent de souris |
| âge | L’âge de la souris (toutes les souris avaient 8 semaines ici) |
| sexe | Le sexe de la souris |
| infection | L’état d’infection de la souris, c’est-à-dire infectée par la grippe A ou non infectée. |
| souche | La souche grippale A. |
| temps | La durée de l’infection (en jours). |
| tissu | Le tissu utilisé pour l’expérience d’expression génique, c’est-à-dire le cervelet ou la moelle épinière. |
| souris | L’identifiant unique de la souris. |
Nous allons utiliser la fonction R download.file() pour
télécharger le fichier CSV qui contient les données d’expression
génique, et nous utiliserons read.csv() pour charger en
mémoire le contenu du fichier CSV en tant qu’objet de classe
data.frame. Dans la commande download.file, la
première entrée est une chaîne de caractères avec l’URL source. Cette
URL source télécharge un fichier CSV à partir d’un référentiel GitHub.
Le texte après la virgule
(“data/rnaseq.csv”) est la destination du fichier sur votre machine locale . Vous aurez besoin d'un dossier sur votre ordinateur appelé“data”dans lequel vous téléchargerez le fichier. Cette commande télécharge donc le fichier distant , le nomme“rnaseq.csv”et l'ajoute à un dossier préexistant nommé“data”`.
R
download.file(url = "https://github.com/carpentries-incubator/bioc-intro/raw/main/episodes/data/rnaseq.csv",
destfile = "data/rnaseq.csv" )
Vous êtes maintenant prêt à charger les données :
R
arn <- read.csv("data/rnaseq.csv")
Cette instruction ne produit aucune sortie car, comme vous vous en souvenez peut-être , les affectations n’affichent rien. Si nous voulons vérifier que nos données ont été chargées, nous pouvons voir le contenu du bloc de données en en tapant son nom :
R
arn
Ouah… c’était beaucoup de production. Au moins, cela signifie que les
données ont été chargées correctement. Vérifions le haut (les 6
premières lignes) de ce bloc de données en utilisant la fonction
head() :
R
head(rna)
ERREUR
Error in eval(expr, envir, enclos): object 'rna' not foundR
## Essayez aussi
## View(rna)
Note
read.csv() suppose que les champs sont délimités par des
virgules, cependant, dans plusieurs pays, la virgule est utilisée comme
séparateur décimal et le point-virgule (;) est utilisé comme champ
délinéateur. Si vous souhaitez lire en ce type de fichiers dans R, vous
pouvez utiliser la fonction read.csv2(). Il se comporte
exactement comme read.csv() mais utilise des paramètres
différents pour la décimale et les séparateurs de champ. Si vous
travaillez avec un autre format , ils peuvent tous deux être spécifiés
par l’utilisateur. Consultez l’aide pour read.csv() en
tapant ?read.csv pour en savoir plus. Il existe également
la fonction read.delim() pour lire des fichiers de données
séparés par des tabulations. Il est important de noter que toutes ces
fonctions sont en fait des fonctions wrapper pour la fonction principale
read.table() avec différents arguments. En tant que tel,
les données ci-dessus auraient également pu être chargées en utilisant
read.table() avec l’argument de séparation comme
,. Le code est comme suit:
R
rna <- read.table(file = "data/rnaseq.csv",
sep = ",",
header = TRUE)
L’argument d’en-tête doit être défini sur TRUE pour pouvoir lire les
en-têtes car par défaut read.table() a l’argument d’en-tête
défini sur FALSE.
Que sont les trames de données ?
Les trames de données sont la structure de données de facto pour la plupart des données tabulaires, et ce que nous utilisons pour les statistiques et le traçage.
Un bloc de données peut être créé à la main, mais le plus souvent ils
sont générés par les fonctions read.csv() ou
read.table() ; en d’autres termes , lors de l’importation
de feuilles de calcul depuis votre disque dur (ou le Web).
Une trame de données est la représentation de données sous le format d’un tableau où les colonnes sont des vecteurs qui ont tous la même longueur. Étant donné que les colonnes sont des vecteurs, chaque colonne doit contenir un seul type de données (par exemple, des caractères, des entiers, des facteurs). Par exemple, voici une figure représentant une trame de données comprenant un chiffre, un caractère et un vecteur logique .

Nous pouvons le voir lors de l’inspection de la structure
d’une trame de données avec la fonction str() :
R
str(arn)
SORTIE
'data.frame': 32428 obs. of 19 variables:
$ gene : chr "Asl" "Apod" "Cyp2d22" "Klk6" ...
$ sample : chr "GSM2545336" "GSM2545336" "GSM2545336" "GSM2545336" ...
$ expression : int 1170 36194 4060 287 85 782 1619 288 43217 1071 ...
$ organism : chr "Mus musculus" "Mus musculus" "Mus musculus" "Mus musculus" ...
$ age : int 8 8 8 8 8 8 8 8 8 8 ...
$ sex : chr "Female" "Female" "Female" "Female" ...
$ infection : chr "InfluenzaA" "InfluenzaA" "InfluenzaA" "InfluenzaA" ...
$ strain : chr "C57BL/6" "C57BL/6" "C57BL/6" "C57BL/6" ...
$ time : int 8 8 8 8 8 8 8 8 8 8 ...
$ tissue : chr "Cerebellum" "Cerebellum" "Cerebellum" "Cerebellum" ...
$ mouse : int 14 14 14 14 14 14 14 14 14 14 ...
$ ENTREZID : int 109900 11815 56448 19144 80891 20528 97827 118454 18823 14696 ...
$ product : chr "argininosuccinate lyase, transcript variant X1" "apolipoprotein D, transcript variant 3" "cytochrome P450, family 2, subfamily d, polypeptide 22, transcript variant 2" "kallikrein related-peptidase 6, transcript variant 2" ...
$ ensembl_gene_id : chr "ENSMUSG00000025533" "ENSMUSG00000022548" "ENSMUSG00000061740" "ENSMUSG00000050063" ...
$ external_synonym : chr "2510006M18Rik" NA "2D22" "Bssp" ...
$ chromosome_name : chr "5" "16" "15" "7" ...
$ gene_biotype : chr "protein_coding" "protein_coding" "protein_coding" "protein_coding" ...
$ phenotype_description : chr "abnormal circulating amino acid level" "abnormal lipid homeostasis" "abnormal skin morphology" "abnormal cytokine level" ...
$ hsapiens_homolog_associated_gene_name: chr "ASL" "APOD" "CYP2D6" "KLK6" ...Inspection des objets data.frame
Nous avons déjà vu comment les fonctions head() et
str() peuvent être utiles pour vérifier le contenu et la
structure d’une trame de données. Voici une liste non exhaustive de
fonctions pour avoir une idée du contenu/structure des données.
Essayons-les !
Taille:
-
dim(rna)- renvoie un vecteur avec le nombre de lignes comme premier élément et le nombre de colonnes comme deuxième élément (les dimensions de l’objet ). -
nrow(rna)- renvoie le nombre de lignes. -
ncol(rna)- renvoie le nombre de colonnes.
Contenu:
-
head(rna)- affiche les 6 premières lignes. -
tail(rna)- affiche les 6 dernières lignes.
Des noms:
-
names(rna)- renvoie les noms de colonnes (synonyme decolnames()pour les objetsdata.frame). -
rownames(rna)- renvoie les noms de lignes.
Résumé:
-
str(rna)- structure de l’objet et informations sur la classe , longueur et contenu de chaque colonne. -
summary(rna)- statistiques récapitulatives pour chaque colonne.
Remarque : la plupart de ces fonctions sont “génériques”, elles
peuvent être utilisées sur d’autres types d’objets en plus de
data.frame.
- classe : trame de données
- combien de lignes : 66465, combien de colonnes : 11
Indexation et sous-ensemble de trames de données
Notre bloc de données « rna » comporte des lignes et des colonnes (il a 2 dimensions) ; si nous voulons en extraire des données spécifiques, nous devons spécifier les “coordonnées” que nous voulons. Les numéros de ligne viennent en premier, suivis des numéros de colonne . Cependant, notez que différentes manières de spécifier ces coordonnées conduisent à des résultats avec des classes différentes.
R
# premier élément de la première colonne du bloc de données (sous forme de vecteur)
rna[1, 1]
# premier élément de la 6ème colonne (sous forme de vecteur)
rna [1, 6]
# première colonne du bloc de données (sous forme de vecteur)
rna[, 1]
# première colonne du bloc de données (sous forme de data.frame )
rna[1]
# les trois premiers éléments de la 7ème colonne (en tant que vecteur)
rna[1:3, 7]
# la 3ème ligne de la trame de données (en tant que data.frame)
rna[3, ]
# équivalent à head_rna <- head(rna)
head_rna <- rna[1:6, ]
head_rna
: est une fonction spéciale qui crée des vecteurs
numériques d’entiers dans ordre croissant ou décroissant, testez
1:10 et 10:1 pour l’instance . Voir la section
@ref(sec:genvec) pour plus de détails.
Vous pouvez également exclure certains indices d’un bloc de données à
l’aide du signe “-” :
R
rna[, -1] ## La trame de données entière, sauf la première colonne
rna[-c(7:66465), ] ## Équivalent à head(rna)
Les trames de données peuvent être sous-ensembles en appelant des indices (comme indiqué précédemment), mais aussi en appelant directement leurs noms de colonnes :
R
rna["gene"] # Result is a data.frame
rna[, "gene"] # Result is a vector
rna[["gene"]] # Result is a vector
rna$gene # Result is a vector
Dans RStudio, vous pouvez utiliser la fonction de saisie semi-automatique pour obtenir les noms complets et corrects des colonnes.
Défi
Créez un
data.frame(rna_200) contenant uniquement les données de ligne 200 de l’ensemble de donnéesrna.Remarquez comment
nrow()vous a donné le nombre de lignes dans undata.frame?
Utilisez ce numéro pour extraire uniquement la dernière ligne de la trame de données initiale
rna.Comparez cela avec ce que vous voyez comme la dernière ligne en utilisant
tail()pour assurez-vous qu’il répond aux attentes.Retirez cette dernière ligne en utilisant
nrow()au lieu du numéro de ligne.Créez un nouveau bloc de données (
rna_last) à partir de cette dernière ligne.
Utilisez
nrow()pour extraire la ligne qui se trouve au milieu du dataframerna. Stockez le contenu de cette ligne dans un objet nommérna_middle.Combinez
nrow()avec la notation-ci-dessus pour reproduire le comportement dehead(rna), en ne conservant que la première à la 6ème lignes de l’ensemble de données rna.
R
## 1.
rna_200 <- rna[200, ]
## 2.
## Sauvegarde de `n_rows` pour améliorer la lisibilité et réduire la duplication
n_rows < - nrow(rna)
ERREUR
Error in eval(expr, envir, enclos): object 'n_rows' not foundR
rna_last <- rna[n_rows, ]
ERREUR
Error in eval(expr, envir, enclos): object 'n_rows' not foundR
## 3.
rna_middle <- rna[n_rows / 2, ]
ERREUR
Error in eval(expr, envir, enclos): object 'n_rows' not foundR
## 4 .
rna_head <- rna[-(7:n_rows), ]
ERREUR
Error in eval(expr, envir, enclos): object 'n_rows' not foundFacteurs
Les facteurs représentent des données catégorielles. Ils sont stockés sous forme d’entiers associés aux étiquettes et ils peuvent être ordonnés ou non. Alors que les facteurs ressemblent (et se comportent souvent) à des vecteurs de caractères, ils sont en fait traités comme des vecteurs entiers par R. Vous devez donc être très prudent lorsque vous les traitez comme des chaînes.
Une fois créés, les facteurs ne peuvent contenir qu’un ensemble prédéfini de valeurs, appelées niveaux. Par défaut, R trie toujours les niveaux par ordre alphabétique . Par exemple, si vous avez un facteur à 2 niveaux :
R
sexe <- factor(c("mâle", "femelle", "femelle", "mâle", "femelle"))
R will assign 1 to the level "female" and
2 to the level "male" (because f
comes before m, even though the first element in this
vector is "male"). Vous pouvez le voir en utilisant la
fonction levels() et vous pouvez trouver le nombre de
niveaux en utilisant nlevels() :
R
niveaux(sexe)
ERREUR
Error in niveaux(sexe): could not find function "niveaux"R
nniveaux(sexe)
ERREUR
Error in nniveaux(sexe): could not find function "nniveaux"Parfois, l’ordre des facteurs n’a pas d’importance, d’autres fois vous souhaiterez peut-être spécifier l’ordre car il est significatif (par exemple, “faible”, “moyen”, “élevé”), il améliore votre visualisation, ou il est requis par un type particulier d’analyse. Ici, une façon de réorganiser nos niveaux dans le vecteur « sexe » serait :
R
sex ## commande actuelle
ERREUR
Error in eval(expr, envir, enclos): object 'sex' not foundR
sex <- factor(sex,levels = c("male", "female"))
ERREUR
Error in eval(expr, envir, enclos): object 'sex' not foundR
sex ## après la nouvelle commande
ERREUR
Error in eval(expr, envir, enclos): object 'sex' not foundDans la mémoire de R, ces facteurs sont représentés par des entiers
(1, 2, 3), mais sont plus informatifs que les entiers car les facteurs
sont auto-descriptifs : "femelle", "mâle" est
plus descriptif que 1, 2. Lequel est
« mâle » ? Vous ne seriez pas en mesure de le savoir uniquement à partir
des données entières . Les facteurs, en revanche, intègrent cette
information. Ceci est particulièrement utile lorsqu’il existe de
nombreux niveaux (comme le biotype du gène dans notre exemple d’ensemble
de données).
Lorsque vos données sont stockées sous forme de facteur, vous pouvez
utiliser la fonction plot() pour avoir un aperçu rapide du
nombre d’observations représenté par chaque niveau de facteur. Regardons
le nombre d’hommes et de femmes dans nos données.
R
intrigue (sexe)
ERREUR
Error in intrigue(sexe): could not find function "intrigue"Conversion en personnage
Si vous devez convertir un facteur en vecteur de caractères, vous
utilisez as.character(x).
R
as.personnage (sexe)
ERREUR
Error in as.personnage(sexe): could not find function "as.personnage"Facteurs de renommage
Si l’on veut renommer ces facteurs, il suffit de changer ses niveaux :
R
niveaux(sexe)
ERREUR
Error in niveaux(sexe): could not find function "niveaux"R
niveaux(sexe) <- c("M", "F")
ERREUR
Error in niveaux(sexe) <- c("M", "F"): could not find function "niveaux<-"R
sexe
SORTIE
[1] mâle femelle femelle mâle femelle
Levels: femelle mâleR
intrigue(sexe)
ERREUR
Error in intrigue(sexe): could not find function "intrigue"Défi:
- Renommez « F » et « M » respectivement en « Femme » et « Mâle ».
R
niveaux(sexe)
ERREUR
Error in niveaux(sexe): could not find function "niveaux"R
niveaux(sexe) <- c("Homme", "Femme")
ERREUR
Error in niveaux(sexe) <- c("Homme", "Femme"): could not find function "niveaux<-"Défi:
Nous avons vu comment les trames de données sont créées lors de
l’utilisation de read.csv(), mais elles peuvent également
être créées à la main avec la fonction data.frame(). Il y a
quelques erreurs dans ce « data.frame » fabriqué à la main. Pouvez-vous
les repérer et les réparer ? N’hésitez pas à expérimenter !
- guillemets manquants autour des noms des animaux
- il manque une entrée dans la colonne “sensation” (probablement pour l’un des animaux à fourrure)
- il manque une virgule dans la colonne poids
Défi:
Pouvez-vous prédire la classe de chacune des colonnes dans l’exemple suivant ?
Vérifiez vos suppositions en utilisant
str(country_climate) :
Sont-ils ce à quoi vous vous attendiez ? Pourquoi? Pourquoi pas?
Réessayez en ajoutant
stringsAsFactors = TRUEaprès la dernière variable lors de la création du bloc de données. Qu’est ce qu’il se passe maintenant?stringsAsFactorspeut également être défini lors de la lecture de feuilles de calcul basées sur du texte dans R à l’aide deread.csv().
R
country_climate <- data.frame(
country = c("Canada", "Panama", "Afrique du Sud", "Australie"),
climat = c("froid", "chaud" , "tempéré", "chaud/tempéré"),
température = c(10, 30, 18, "15"),
hémisphère_nord = c(VRAI, VRAI, FAUX, "FAUX" ),
has_kangaroo = c(FALSE, FALSE, FALSE, 1)
)
R
country_climate <- data.frame(
country = c("Canada", "Panama", "Afrique du Sud", "Australie"),
climat = c("froid", "chaud" , "tempéré", "chaud/tempéré"),
température = c(10, 30, 18, "15"),
hémisphère_nord = c(VRAI, VRAI, FAUX, "FAUX" ),
has_kangaroo = c(FALSE, FALSE, FALSE, 1)
)
ERREUR
Error in eval(expr, envir, enclos): object 'VRAI' not foundR
str(country_climate)
ERREUR
Error in eval(expr, envir, enclos): object 'country_climate' not foundLa conversion automatique du type de données est parfois une bénédiction, parfois un désagrément. Sachez qu’il existe, apprenez les règles et vérifiez que les données que vous importez dans R sont du type correct dans votre bloc de données. Sinon, utilisez-le à votre avantage pour détecter les erreurs qui auraient pu être introduites lors de la saisie des données (une lettre dans une colonne qui ne doit contenir que des chiffres par exemple).
Apprenez-en plus dans ce tutoriel RStudio
Matrices
Avant de continuer, maintenant que nous avons découvert les trames de
données, récapitulons l’installation du package et découvrons un nouveau
type de données, à savoir la matrice. Comme un
data.frame, une matrice a deux dimensions, des lignes et
colonnes. Mais la différence majeure est que toutes les cellules d’une «
matrice » doivent être du même type : « numérique », « caractère », «
logique », … À cet égard , les matrices sont plus proches d’un « vecteur
» que d’un « data.frame ».
Le constructeur par défaut d’une matrice est « matrice ». Il faut un vecteur de valeurs pour remplir la matrice et le nombre de lignes et/ou colonnes1. Les valeurs sont triées le long des colonnes, comme illustré ci-dessous.
R
m <- matrice (1:9, ncol = 3, nrow = 3)
ERREUR
Error in matrice(1:9, ncol = 3, nrow = 3): could not find function "matrice"R
m
ERREUR
Error in eval(expr, envir, enclos): object 'm' not foundIl est souvent utile de créer de grandes matrices de données
aléatoires comme données de test . L’exercice ci-dessous vous demande de
créer une telle matrice avec des données aléatoires tirées d’une
distribution normale de moyenne 0 et d’écart type 1, ce qui peut être
fait avec la fonction rnorm().
R
set.seed(123)
m <- matrice(rnorm(3000), ncol = 3)
ERREUR
Error in matrice(rnorm(3000), ncol = 3): could not find function "matrice"R
dim(m)
ERREUR
Error in eval(expr, envir, enclos): object 'm' not foundR
tête(m)
ERREUR
Error in tête(m): could not find function "tête"Formatage des dates
L’un des problèmes les plus courants rencontrés par les nouveaux (et expérimentés !) Les utilisateurs de R ont la conversion des informations de date et d’heure en une variable appropriée et utilisable lors des analyses.
Remarque sur les dates dans les tableurs
Les dates dans les feuilles de calcul sont généralement stockées dans une seule colonne. Bien que cela semble la manière la plus naturelle d’enregistrer des dates, ce n’est en réalité pas la meilleure pratique. Un tableur affichera les dates d’une manière apparemment correcte (pour un observateur humain), mais la façon dont elle gère et stocke réellement les dates peut être problématique. Il est souvent plus sûr de stocker les dates avec ANNÉE, MOIS et JOUR dans des colonnes séparées ou comme ANNÉE et JOUR DE L’ANNÉE dans des colonnes séparées.
Tableurs tels que LibreOffice, Microsoft Excel, OpenOffice, Gnumeric, … ont des manières différentes (et souvent incompatibles) d’encoder les dates (même pour le même programme entre les versions et les systèmes d’exploitation ). De plus, Excel peut transformer des éléments qui ne sont pas des dates en dates (@Zeeberg:2004), par exemple des noms ou des identifiants comme MAR1, DEC1, OCT4. Donc, si vous évitez globalement le format de date, il est plus facile d’ identifier ces problèmes.
La section Dates as data de la leçon Data Carpentry fournit des informations supplémentaires sur les pièges des dates avec des feuilles de calcul.
Nous allons utiliser la fonction ymd() du package
lubridate (qui appartient au
tidyverse ; en savoir plus [ici] (https://www.tidyverse.org/)). .
lubridate est installé dans le cadre de
l’installation de tidyverse. Lorsque vous
chargez le tidyverse
(library(tidyverse)), les packages de base (les packages
utilisés dans la plupart des analyses de données) sont chargés.
lubridate n’appartient cependant pas au
noyau spiceverse, vous devez donc le charger explicitement avec
library(lubridate).
Commencez par charger le package requis :
R
bibliothèque("lubrifier")
ERREUR
Error in bibliothèque("lubrifier"): could not find function "bibliothèque"ymd() prend un vecteur représentant l’année, le mois et
le jour, et le convertit en un vecteur Date.
Date est une classe de données reconnue par R comme étant
une date et peut être manipulée comme telle. L’argument requis par la
fonction est flexible, mais, à titre de bonne pratique, il s’agit d’un
vecteur de caractère au format “AAAA-MM-JJ”.
Créons un objet date et inspectons la structure :
R
ma_date <- ymd("2015-01-01")
ERREUR
Error in ymd("2015-01-01"): could not find function "ymd"R
str(ma_date)
ERREUR
Error in eval(expr, envir, enclos): object 'ma_date' not foundCollons maintenant l’année, le mois et le jour séparément - nous obtenons le même résultat :
R
# sep indique le caractère à utiliser pour séparer chaque composant
my_date <- ymd(paste("2015", "1", "1", sep = "-"))
ERREUR
Error in ymd(paste("2015", "1", "1", sep = "-")): could not find function "ymd"R
str(my_date )
ERREUR
Error in eval(expr, envir, enclos): object 'my_date' not foundFamiliarisons-nous maintenant avec un pipeline typique de manipulation de date . Les petites données ci-dessous ont stocké des dates dans différentes colonnes « année », « mois » et « jour ».
R
x <- data.frame(année = c(1996, 1992, 1987, 1986, 2000, 1990, 2002, 1994, 1997, 1985),
mois = c(2, 3, 3, 10, 1 , 8, 3, 4, 5, 5),
jour = c(24, 8, 1, 5, 8, 17, 13, 10, 11, 24),
valeur = c (4, 5, 1, 9, 3, 8, 10, 2, 6, 7))
x
SORTIE
année mois jour valeur
1 1996 2 24 4
2 1992 3 8 5
3 1987 3 1 1
4 1986 10 5 9
5 2000 1 8 3
6 1990 8 17 8
7 2002 3 13 10
8 1994 4 10 2
9 1997 5 11 6
10 1985 5 24 7Nous appliquons maintenant cette fonction à l’ensemble de données
« x ». Nous créons d’abord un vecteur de caractères à partir des
colonnes year, month et day de
x en utilisant paste() :
R
coller(x$year, x$month, x$day, sep = "-")
ERREUR
Error in coller(x$year, x$month, x$day, sep = "-"): could not find function "coller"Ce vecteur de caractères peut être utilisé comme argument pour
ymd() :
R
ymd(coller(x$year, x$month, x$day, sep = "-"))
ERREUR
Error in ymd(coller(x$year, x$month, x$day, sep = "-")): could not find function "ymd"Le vecteur Date résultant peut être ajouté à
x en tant que nouvelle colonne appelée
date :
R
x$date <- ymd(paste(x$year, x$month, x$day, sep = "-"))
ERREUR
Error in ymd(paste(x$year, x$month, x$day, sep = "-")): could not find function "ymd"R
str(x) # remarquez la nouvelle colonne, avec 'date' comme classe
SORTIE
'data.frame': 10 obs. of 4 variables:
$ année : num 1996 1992 1987 1986 2000 ...
$ mois : num 2 3 3 10 1 8 3 4 5 5
$ jour : num 24 8 1 5 8 17 13 10 11 24
$ valeur: num 4 5 1 9 3 8 10 2 6 7Assurons-nous que tout a fonctionné correctement. Une façon
d’inspecter la nouvelle colonne est d’utiliser
summary() :
R
résumé(x$date)
ERREUR
Error in résumé(x$date): could not find function "résumé"Notez que ymd() s’attend à avoir l’année, le mois et le
jour, dans cet ordre . Si vous avez par exemple le jour, le mois et
l’année, vous aurez besoin de dmy().
R
dmy(coller(x$day, x$month, x$year, sep = "-"))
ERREUR
Error in dmy(coller(x$day, x$month, x$year, sep = "-")): could not find function "dmy"lubdridate a de nombreuses fonctions pour gérer toutes
les variations de date.
Résumé des objets R
Jusqu’à présent, nous avons vu plusieurs types d’objets R variant selon le nombre de dimensions et s’ils pouvaient stocker un ou plusieurs types de données :
-
vecteur: une dimension (ils ont une longueur), un seul type de données. -
matrice: deux dimensions, un seul type de données. -
data.frame: deux dimensions, un type par colonne.
Listes
Un type de données que nous n’avons pas encore vu, mais qu’il est utile de connaître, et découle du résumé que nous venons de voir sont des listes :
-
list: une dimension, chaque élément peut être d’un type de données différent .
Ci-dessous, créons une liste contenant un vecteur de nombres, de caractères, une matrice, un dataframe et une autre liste :
R
l <- list(1:10, ## numérique
lettres, ## caractère
installé.packages(), ## une matrice
voitures, ## un data.frame
liste(1, 2, 3)) ## une liste
ERREUR
Error in eval(expr, envir, enclos): object 'lettres' not foundR
longueur(l)
ERREUR
Error in longueur(l): could not find function "longueur"R
str(l)
ERREUR
Error in eval(expr, envir, enclos): object 'l' not foundLe sous-ensemble de liste est effectué en utilisant []
pour sous-ensembler une nouvelle sous-liste ou [[]] pour
extraire un seul élément de cette liste (en utilisant des indices ou des
noms, si la liste est appelé).
R
l[[1]] ## premier élément
ERREUR
Error in eval(expr, envir, enclos): object 'l' not foundR
l[1:2] ## une liste de longueur 2
ERREUR
Error in eval(expr, envir, enclos): object 'l' not foundR
l[1] ## une liste de longueur 1
ERREUR
Error in eval(expr, envir, enclos): object 'l' not foundExportation et sauvegarde de données tabulaires
Nous avons vu comment lire une feuille de calcul textuelle dans R à
l’aide de la famille de fonctions read.table. Pour exporter
un data.frame vers une feuille de calcul texte , nous
pouvons utiliser l’ensemble de fonctions write.table
(write.csv, write.delim, …). Ils prennent tous
la variable à exportée et le fichier vers lequel exporter. Par exemple,
pour exporter les données rna vers le fichier
my_rna.csv dans le répertoire data_output ,
nous exécuterions :
R
write.csv(rna, file = "data_output/my_rna.csv")
This new csv file can now be shared with other collaborators who
aren’t familiar with R. Note that even though there are commas in some
of the fields in the data.frame (see for example the
“product” column), R will by default surround each field with quotes,
and thus we will be able to read it back into R correctly, despite also
using commas as column separators.
Soit le nombre de lignes, soit le nombre de colonnes sont suffisants, l’autre pouvant être déduit de la longueur des valeurs. Essayez ce qui se passe si les valeurs et le nombre de lignes/colonnes ne s’additionnent pas.↩︎
Content from Manipulation et analyse de données avec dplyr
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 150 minutes
Vue d'ensemble
Questions
- Analyse de données dans R à l’aide du méta-paquet Tidyverse
Objectifs
- Décrivez l’objectif des packages
dplyrettidyr. - Décrivez plusieurs de leurs fonctions extrêmement utiles pour manipuler des données.
- Décrivez le concept d’un format de tableau large et long, et voyez comment remodeler un bloc de données d’un format à l’autre.
- Montrez comment joindre des tables.
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Manipulation des données à l’aide de
dplyr et
tidyr
Le sous-ensemble entre crochets est pratique, mais il peut être fastidieux et difficile à lire, en particulier pour les opérations compliquées.
Certains packages peuvent grandement faciliter notre tâche lorsque
nous manipulons des données. Les packages dans R sont essentiellement
des ensembles de fonctions supplémentaires qui vous permettent de faire
plus de choses. Les fonctions que nous avons utilisées jusqu’à présent,
comme str() ou data.frame(), sont intégrées à
R ; Le chargement de packages peut vous donner accès à d’autres
fonctions spécifiques. Avant d’utiliser un package pour la première
fois, vous devez l’installer sur votre machine, puis vous devez
l’importer à chaque session R suivante lorsque vous en avez besoin.
Le package
dplyrfournit des outils puissants pour les tâches de manipulation de données. Il est conçu pour fonctionner directement avec des trames de données, avec de nombreuses tâches de manipulation optimisées.Comme nous le verrons plus loin, nous souhaitons parfois qu’un bloc de données soit remodelé pour pouvoir effectuer des analyses spécifiques ou pour la visualisation. Le package
tidyrrésout ce problème courant de remodelage des données et fournit des outils pour manipuler les données de manière ordonnée.
Pour en savoir plus sur dplyr et
tidyr après l’atelier, vous voudrez
peut-être consulter ceci transformation de données pratique avec ** et
ceci celui sur .
- Le package
tidyverseest un “package parapluie” qui installe plusieurs packages utiles pour l’analyse des données qui fonctionnent bien ensemble, tels quetidyr, * *dplyr**,ggplot2,tibble, etc. Ces packages nous aident à travailler et à interagir avec les données. Ils nous permettent de faire beaucoup de choses avec vos données, comme le sous-ensemble, la transformation, la visualisation, etc.
Si vous avez effectué la configuration, vous devriez déjà avoir installé le package Tidyverse. Vérifiez si vous l’avez en essayant de le charger depuis la bibliothèque :
R
## load the tidyverse packages, incl. dplyr
library("tidyverse")
Si vous recevez un message d’erreur
il n'y a pas de package appelé 'tidyverse' alors vous
n’avez pas installé le package pour cette version de R. Pour installer
le package tidyverse, tapez :
R
BiocManager::install("tidyverse")
Si vous avez dû installer le package
tidyverse, n’oubliez pas de le charger
dans cette session R en utilisant la commande library()
ci-dessus !
Chargement de données avec Tidyverse
Instead of read.csv(), we will read in our data using
the read_csv() function (notice the _ instead
of the .), from the tidyverse package
readr.
R
rna <- read_csv("data/rnaseq.csv")
## view the data
rna
SORTIE
# A tibble: 32,428 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Notez que la classe des données est désormais appelée « tibble ».
Tibbles modifie certains des comportements des objets de trame de données que nous avons introduits dans le précédemment. La structure des données est très similaire à une trame de données. Pour nos besoins , les seules différences sont les suivantes :
Il affiche le type de données de chaque colonne sous son nom. Notez que <
dbl> est un type de données défini pour contenir des valeurs numériques avec points décimaux.Il imprime uniquement les premières lignes de données et seulement autant de colonnes que peuvent contenir un écran.
Nous allons maintenant apprendre certaines des fonctions
dplyr les plus courantes :
-
select(): sous-ensemble de colonnes -
filter(): sous-ensemble de lignes sur conditions -
mutate(): crée de nouvelles colonnes en utilisant les informations d’autres colonnes -
group_by()etsummarise(): créent des statistiques récapitulatives sur des données groupées -
arrange(): trier les résultats -
count(): compte les valeurs discrètes
Sélection de colonnes et filtrage de lignes
Pour sélectionner les colonnes d’un bloc de données, utilisez
select(). Le premier argument de cette fonction est la
trame de données (rna), et les arguments suivants sont les
colonnes à conserver.
R
select(rna, gene, sample, tissue, expression)
SORTIE
# A tibble: 32,428 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545336 Cerebellum 1170
2 Apod GSM2545336 Cerebellum 36194
3 Cyp2d22 GSM2545336 Cerebellum 4060
4 Klk6 GSM2545336 Cerebellum 287
5 Fcrls GSM2545336 Cerebellum 85
6 Slc2a4 GSM2545336 Cerebellum 782
7 Exd2 GSM2545336 Cerebellum 1619
8 Gjc2 GSM2545336 Cerebellum 288
9 Plp1 GSM2545336 Cerebellum 43217
10 Gnb4 GSM2545336 Cerebellum 1071
# ℹ 32,418 more rowsPour sélectionner toutes les colonnes sauf certaines, mettez un “-” devant la variable pour l’exclure.
R
select(rna, -tissue, -organism)
SORTIE
# A tibble: 32,428 × 17
gene sample expression age sex infection strain time mouse ENTREZID
<chr> <chr> <dbl> <dbl> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Asl GSM2545… 1170 8 Fema… Influenz… C57BL… 8 14 109900
2 Apod GSM2545… 36194 8 Fema… Influenz… C57BL… 8 14 11815
3 Cyp2d22 GSM2545… 4060 8 Fema… Influenz… C57BL… 8 14 56448
4 Klk6 GSM2545… 287 8 Fema… Influenz… C57BL… 8 14 19144
5 Fcrls GSM2545… 85 8 Fema… Influenz… C57BL… 8 14 80891
6 Slc2a4 GSM2545… 782 8 Fema… Influenz… C57BL… 8 14 20528
7 Exd2 GSM2545… 1619 8 Fema… Influenz… C57BL… 8 14 97827
8 Gjc2 GSM2545… 288 8 Fema… Influenz… C57BL… 8 14 118454
9 Plp1 GSM2545… 43217 8 Fema… Influenz… C57BL… 8 14 18823
10 Gnb4 GSM2545… 1071 8 Fema… Influenz… C57BL… 8 14 14696
# ℹ 32,418 more rows
# ℹ 7 more variables: product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>Cela sélectionnera toutes les variables de rna sauf
tissu et organism.
Pour choisir des lignes en fonction d’un critère spécifique, utilisez
filter() :
R
filter(rna, sex == "Male")
SORTIE
# A tibble: 14,740 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 626 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
2 Apod GSM254… 13021 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
3 Cyp2d22 GSM254… 2171 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
4 Klk6 GSM254… 448 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
5 Fcrls GSM254… 180 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
6 Slc2a4 GSM254… 313 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
7 Exd2 GSM254… 2366 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
8 Gjc2 GSM254… 310 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
9 Plp1 GSM254… 53126 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
10 Gnb4 GSM254… 1355 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
# ℹ 14,730 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>R
filter(rna, sex == "Male" & infection == "NonInfected")
SORTIE
# A tibble: 4,422 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 535 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
2 Apod GSM254… 13668 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
3 Cyp2d22 GSM254… 2008 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
4 Klk6 GSM254… 1101 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
5 Fcrls GSM254… 375 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
6 Slc2a4 GSM254… 249 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
7 Exd2 GSM254… 3126 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
8 Gjc2 GSM254… 791 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
9 Plp1 GSM254… 98658 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
10 Gnb4 GSM254… 2437 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
# ℹ 4,412 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Imaginons maintenant que nous nous intéressions aux homologues
humains des gènes de souris analysés dans cet ensemble de données. Ces
informations se trouvent dans la dernière colonne du tibble
rna, nommée
hsapiens_homolog_associated_gene_name. Pour le visualiser
facilement, nous allons créer un nouveau tableau contenant uniquement
les 2 colonnes gene et
hsapiens_homolog_associated_gene_name.
R
genes <- select(rna, gene, hsapiens_homolog_associated_gene_name)
genes
SORTIE
# A tibble: 32,428 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Asl ASL
2 Apod APOD
3 Cyp2d22 CYP2D6
4 Klk6 KLK6
5 Fcrls FCRL2
6 Slc2a4 SLC2A4
7 Exd2 EXD2
8 Gjc2 GJC2
9 Plp1 PLP1
10 Gnb4 GNB4
# ℹ 32,418 more rowsCertains gènes de souris n’ont pas d’homologues humains. Ceux-ci
peuvent être récupérés en utilisant filter() et la fonction
is.na(), qui détermine si quelque chose est un
NA.
R
filter(genes, is.na(hsapiens_homolog_associated_gene_name))
SORTIE
# A tibble: 4,290 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Prodh <NA>
2 Tssk5 <NA>
3 Vmn2r1 <NA>
4 Gm10654 <NA>
5 Hexa <NA>
6 Sult1a1 <NA>
7 Gm6277 <NA>
8 Tmem198b <NA>
9 Adam1a <NA>
10 Ebp <NA>
# ℹ 4,280 more rowsSi on veut conserver uniquement les gènes de souris qui ont un
homologue humain, on peut insérer un “!” symbole qui annule le résultat,
nous demandons donc chaque ligne où
hsapiens_homolog_associated_gene_name n’est pas un
NA.
R
filter(genes, !is.na(hsapiens_homolog_associated_gene_name))
SORTIE
# A tibble: 28,138 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Asl ASL
2 Apod APOD
3 Cyp2d22 CYP2D6
4 Klk6 KLK6
5 Fcrls FCRL2
6 Slc2a4 SLC2A4
7 Exd2 EXD2
8 Gjc2 GJC2
9 Plp1 PLP1
10 Gnb4 GNB4
# ℹ 28,128 more rowsTuyaux
Et si vous souhaitez sélectionner et filtrer en même temps ? Il existe trois façons de procéder : utiliser des étapes intermédiaires, des fonctions imbriquées ou des tuyaux.
Avec des étapes intermédiaires, vous créez un bloc de données temporaire et l’utilisez comme entrée de la fonction suivante, comme ceci :
R
rna2 <- filter(rna, sex == "Male")
rna3 <- select(rna2, gene, sample, tissue, expression)
rna3
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsCeci est lisible, mais peut encombrer votre espace de travail avec de nombreux objets intermédiaires que vous devez nommer individuellement. Avec plusieurs étapes , cela peut être difficile à suivre.
Vous pouvez également imbriquer des fonctions (c’est-à-dire une fonction dans une autre), comme ceci :
R
rna3 <- select(filter(rna, sex == "Male"), gene, sample, tissue, expression)
rna3
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsC’est pratique, mais peut être difficile à lire si trop de fonctions sont imbriquées, car R évalue l’expression de l’intérieur vers l’extérieur (dans ce cas, filtrer, puis sélectionner).
La dernière option, pipes, est un ajout récent à R. Pipes vous permet de prendre la sortie d’une fonction et de l’envoyer directement à la suivante, ce qui est utile lorsque vous devez faire beaucoup de choses dans le même ensemble de données.
Les tuyaux dans R ressemblent à %>% (mis à
disposition via le package magrittr ) ou
|> (via la base R). If you use RStudio, you can type the
pipe with Ctrl + Shift + M if you have
a PC or Cmd + Shift + M if you have a
Mac.
Dans le code ci-dessus, nous utilisons le tube pour envoyer
l’ensemble de données rna d’abord via filter()
pour conserver les lignes où sex est Homme, puis via
select() pour conserver uniquement les colonnes
gène, échantillon, tissu et
expression.
Le tube %>% prend l’objet à sa gauche et le passe
directement comme le premier argument de la fonction à sa droite, nous
n’avons pas besoin de inclure explicitement le bloc de données comme un
argument pour les fonctions filter() et
select().
R
rna %>%
filter(sex == "Male") %>%
select(gene, sample, tissue, expression)
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsCertains trouveront peut-être utile de lire le tube comme le mot «
alors ». Par exemple, dans l’exemple ci-dessus, nous avons pris la trame
de données rna, puis nous avons
filtré pour les lignes avec sex == "Male",
puis nous avons « sélectionné les colonnes « gène », «
échantillon », « tissu » et « expression ».
Les fonctions dplyr en elles-mêmes sont
quelque peu simples, mais en les combinant dans des flux de travail
linéaires avec le tube, nous pouvons accomplir des manipulations plus
complexes de trames de données.
Si nous voulons créer un nouvel objet avec cette version plus petite des données, nous pouvons lui attribuer un nouveau nom :
R
rna3 <- rna %>%
filter(sex == "Male") %>%
select(gene, sample, tissue, expression)
rna3
SORTIE
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsR
rna %>%
filter(expression > 50000,
sex == "Female",
time == 0 ) %>%
select(gene, sample, time, expression, age)
SORTIE
# A tibble: 9 × 5
gene sample time expression age
<chr> <chr> <dbl> <dbl> <dbl>
1 Plp1 GSM2545337 0 101241 8
2 Atp1b1 GSM2545337 0 53260 8
3 Plp1 GSM2545338 0 96534 8
4 Atp1b1 GSM2545338 0 50614 8
5 Plp1 GSM2545348 0 102790 8
6 Atp1b1 GSM2545348 0 59544 8
7 Plp1 GSM2545353 0 71237 8
8 Glul GSM2545353 0 52451 8
9 Atp1b1 GSM2545353 0 61451 8Subir une mutation
Vous souhaiterez fréquemment créer de nouvelles colonnes basées sur
les valeurs des colonnes existantes, par exemple pour effectuer des
conversions d’unités ou pour trouver le rapport des valeurs dans deux
colonnes . Pour cela, nous utiliserons mutate().
Pour créer une nouvelle colonne de temps en heures :
R
rna %>%
mutate(time_hours = time * 24) %>%
select(time, time_hours)
SORTIE
# A tibble: 32,428 × 2
time time_hours
<dbl> <dbl>
1 8 192
2 8 192
3 8 192
4 8 192
5 8 192
6 8 192
7 8 192
8 8 192
9 8 192
10 8 192
# ℹ 32,418 more rowsVous pouvez également créer une deuxième nouvelle colonne basée sur
la première nouvelle colonne dans le même appel de
mutate() :
R
rna %>%
mutate(time_hours = time * 24,
time_mn = time_hours * 60) %>%
select(time, time_hours, time_mn)
SORTIE
# A tibble: 32,428 × 3
time time_hours time_mn
<dbl> <dbl> <dbl>
1 8 192 11520
2 8 192 11520
3 8 192 11520
4 8 192 11520
5 8 192 11520
6 8 192 11520
7 8 192 11520
8 8 192 11520
9 8 192 11520
10 8 192 11520
# ℹ 32,418 more rowsDéfi
Créez un nouveau bloc de données à partir des données
rna qui répond aux critères suivants : contient uniquement
le gène, le nom_chromosome,
phenotype_description, sample et
expression Colonnes. Les valeurs de l’expression doivent
être transformées en log. Cette trame de données doit contenir
uniquement des gènes situés sur les chromosomes sexuels, associés à un
phénotype _description, et avec une expression log supérieure à 5.
Astuce : réfléchissez à la façon dont les commandes doivent être ordonnées pour produire ce bloc de données !
R
rna %>%
mutate(expression = log(expression)) %>%
select(gene, chromosome_name, phenotype_description, sample, expression) %>%
filter(chromosome_name == "X" | chromosome_name == "Y") %>%
filter(!is.na(phenotype_description)) %>%
filter(expression > 5)
SORTIE
# A tibble: 649 × 5
gene chromosome_name phenotype_description sample expression
<chr> <chr> <chr> <chr> <dbl>
1 Plp1 X abnormal CNS glial cell morphology GSM25… 10.7
2 Slc7a3 X decreased body length GSM25… 5.46
3 Plxnb3 X abnormal coat appearance GSM25… 6.58
4 Rbm3 X abnormal liver morphology GSM25… 9.32
5 Cfp X abnormal cardiovascular system phys… GSM25… 6.18
6 Ebp X abnormal embryonic erythrocyte morp… GSM25… 6.68
7 Cd99l2 X abnormal cellular extravasation GSM25… 8.04
8 Piga X abnormal brain development GSM25… 6.06
9 Pim2 X decreased T cell proliferation GSM25… 7.11
10 Itm2a X no abnormal phenotype detected GSM25… 7.48
# ℹ 639 more rowsAnalyse de données fractionnée-appliquée-combinée
De nombreuses tâches d’analyse de données peuvent être abordées à
l’aide du paradigme split-apply-combine : divisez les données
en groupes, appliquez une analyse à chaque groupe, puis combinez les
résultats. dplyr rend cela très facile
grâce à l’utilisation de la fonction group_by().
R
rna %>%
group_by(gene)
SORTIE
# A tibble: 32,428 × 19
# Groups: gene [1,474]
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>La fonction group_by() n’effectue aucun traitement de
données, elle regroupe les données en sous-ensembles : dans l’exemple
ci-dessus, notre tibble initial de 32428 les observations
sont divisées en 1474 en fonction de la variable gene.
On pourrait de même décider de regrouper les tibbles par échantillons :
R
rna %>%
group_by(sample)
SORTIE
# A tibble: 32,428 × 19
# Groups: sample [22]
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Ici, notre tibble initial d’observations 32428 est
divisé en groupes 22 en fonction de la variable sample.
Une fois les données regroupées, les opérations suivantes seront appliquées sur chaque groupe indépendamment.
La fonction summaris()
group_by() est souvent utilisé avec
summarise(), qui réduit chaque groupe en un résumé sur une
seule ligne de ce groupe.
group_by() prend comme arguments les noms de colonnes
qui contiennent les variables catégorielles pour
lesquelles vous souhaitez calculer les statistiques récapitulatives .
Donc, pour calculer l’expression moyenne par gène :
R
rna %>%
group_by(gene) %>%
summarise(mean_expression = mean(expression))
SORTIE
# A tibble: 1,474 × 2
gene mean_expression
<chr> <dbl>
1 AI504432 1053.
2 AW046200 131.
3 AW551984 295.
4 Aamp 4751.
5 Abca12 4.55
6 Abcc8 2498.
7 Abhd14a 525.
8 Abi2 4909.
9 Abi3bp 1002.
10 Abl2 2124.
# ℹ 1,464 more rowsNous pourrions également vouloir calculer les niveaux d’expression moyens de tous les gènes dans chaque échantillon :
R
rna %>%
group_by(sample) %>%
summarise(mean_expression = mean(expression))
SORTIE
# A tibble: 22 × 2
sample mean_expression
<chr> <dbl>
1 GSM2545336 2062.
2 GSM2545337 1766.
3 GSM2545338 1668.
4 GSM2545339 1696.
5 GSM2545340 1682.
6 GSM2545341 1638.
7 GSM2545342 1594.
8 GSM2545343 2107.
9 GSM2545344 1712.
10 GSM2545345 1700.
# ℹ 12 more rowsMais on peut aussi regrouper par plusieurs colonnes :
R
rna %>%
group_by(gene, infection, time) %>%
summarise(mean_expression = mean(expression))
SORTIE
`summarise()` has grouped output by 'gene', 'infection'. You can override using
the `.groups` argument.SORTIE
# A tibble: 4,422 × 4
# Groups: gene, infection [2,948]
gene infection time mean_expression
<chr> <chr> <dbl> <dbl>
1 AI504432 InfluenzaA 4 1104.
2 AI504432 InfluenzaA 8 1014
3 AI504432 NonInfected 0 1034.
4 AW046200 InfluenzaA 4 152.
5 AW046200 InfluenzaA 8 81
6 AW046200 NonInfected 0 155.
7 AW551984 InfluenzaA 4 302.
8 AW551984 InfluenzaA 8 342.
9 AW551984 NonInfected 0 238
10 Aamp InfluenzaA 4 4870
# ℹ 4,412 more rowsUne fois les données regroupées, vous pouvez également résumer plusieurs variables en même temps (et pas nécessairement sur la même variable). Par exemple, nous pourrions ajouter une colonne indiquant l’expression médiane par gène et par condition :
R
rna %>%
group_by(gene, infection, time) %>%
summarise(mean_expression = mean(expression),
median_expression = median(expression))
SORTIE
`summarise()` has grouped output by 'gene', 'infection'. You can override using
the `.groups` argument.SORTIE
# A tibble: 4,422 × 5
# Groups: gene, infection [2,948]
gene infection time mean_expression median_expression
<chr> <chr> <dbl> <dbl> <dbl>
1 AI504432 InfluenzaA 4 1104. 1094.
2 AI504432 InfluenzaA 8 1014 985
3 AI504432 NonInfected 0 1034. 1016
4 AW046200 InfluenzaA 4 152. 144.
5 AW046200 InfluenzaA 8 81 82
6 AW046200 NonInfected 0 155. 163
7 AW551984 InfluenzaA 4 302. 245
8 AW551984 InfluenzaA 8 342. 287
9 AW551984 NonInfected 0 238 265
10 Aamp InfluenzaA 4 4870 4708
# ℹ 4,412 more rowsR
rna %>%
filter(gene == "Dok3") %>%
group_by(time) %>%
summarise(mean = mean(expression))
SORTIE
# A tibble: 3 × 2
time mean
<dbl> <dbl>
1 0 169
2 4 156.
3 8 61 Compte
Lorsque nous travaillons avec des données, nous souhaitons souvent
connaître le nombre d’observations trouvées pour chaque facteur ou
combinaison de facteurs. Pour cette tâche,
dplyr fournit count(). Par
exemple, si nous voulions compter le nombre de lignes de données pour
chaque échantillon infecté et non infecté, nous ferions :
R
rna %>%
count(infection)
SORTIE
# A tibble: 2 × 2
infection n
<chr> <int>
1 InfluenzaA 22110
2 NonInfected 10318La fonction count() est un raccourci pour quelque chose
que nous avons déjà vu : regrouper par une variable et le résumer en
comptant le nombre d’observations dans ce groupe. En d’autres termes,
« rna %>% count(infection) » équivaut à :
R
rna %>%
group_by(infection) %>%
summarise(n = n())
SORTIE
# A tibble: 2 × 2
infection n
<chr> <int>
1 InfluenzaA 22110
2 NonInfected 10318L’exemple précédent montre l’utilisation de count() pour
compter le nombre de lignes/observations pour un facteur
(c’est-à-dire infection). Si nous voulions compter une
combinaison de facteurs, telle que infection et
time, nous spécifierions le premier et le deuxième facteur
comme arguments de count() :
R
rna %>%
count(infection, time)
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318ce qui équivaut à ceci :
R
rna %>%
group_by(infection, time) %>%
summarise(n = n())
SORTIE
`summarise()` has grouped output by 'infection'. You can override using the
`.groups` argument.SORTIE
# A tibble: 3 × 3
# Groups: infection [2]
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318Il est parfois utile de trier le résultat pour faciliter les
comparaisons. Nous pouvons utiliser arrange() pour trier le
tableau. Par exemple, nous pourrions vouloir organiser le tableau
ci-dessus par heure :
R
rna %>%
count(infection, time) %>%
arrange(time)
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 NonInfected 0 10318
2 InfluenzaA 4 11792
3 InfluenzaA 8 10318ou par comptages :
R
rna %>%
count(infection, time) %>%
arrange(n)
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 8 10318
2 NonInfected 0 10318
3 InfluenzaA 4 11792Pour trier par ordre décroissant, nous devons ajouter la fonction
desc() :
R
rna %>%
count(infection, time) %>%
arrange(desc(n))
SORTIE
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318Défi
- Combien de gènes ont été analysés dans chaque échantillon ?
- Utilisez
group_by()etsummarise()pour évaluer la profondeur de séquençage (la somme de tous les comptes) dans chaque échantillon. Quel échantillon a la profondeur de séquençage la plus élevée ? - Choisissez un échantillon et évaluez le nombre de gènes par biotype.
- Identifiez les gènes associés à la description du phénotype « méthylation anormale de l’ADN » et calculez leur expression moyenne (en log) au temps 0, au temps 4 et au temps 8.
R
## 1.
rna %>%
count(sample)
SORTIE
# A tibble: 22 × 2
sample n
<chr> <int>
1 GSM2545336 1474
2 GSM2545337 1474
3 GSM2545338 1474
4 GSM2545339 1474
5 GSM2545340 1474
6 GSM2545341 1474
7 GSM2545342 1474
8 GSM2545343 1474
9 GSM2545344 1474
10 GSM2545345 1474
# ℹ 12 more rowsR
## 2.
rna %>%
group_by(sample) %>%
summarise(seq_depth = sum(expression)) %>%
arrange(desc(seq_depth))
SORTIE
# A tibble: 22 × 2
sample seq_depth
<chr> <dbl>
1 GSM2545350 3255566
2 GSM2545352 3216163
3 GSM2545343 3105652
4 GSM2545336 3039671
5 GSM2545380 3036098
6 GSM2545353 2953249
7 GSM2545348 2913678
8 GSM2545362 2913517
9 GSM2545351 2782464
10 GSM2545349 2758006
# ℹ 12 more rowsR
## 3.
rna %>%
filter(sample == "GSM2545336") %>%
count(gene_biotype) %>%
arrange(desc(n))
SORTIE
# A tibble: 13 × 2
gene_biotype n
<chr> <int>
1 protein_coding 1321
2 lncRNA 69
3 processed_pseudogene 59
4 miRNA 7
5 snoRNA 5
6 TEC 4
7 polymorphic_pseudogene 2
8 unprocessed_pseudogene 2
9 IG_C_gene 1
10 scaRNA 1
11 transcribed_processed_pseudogene 1
12 transcribed_unitary_pseudogene 1
13 transcribed_unprocessed_pseudogene 1R
## 4.
rna %>%
filter(phenotype_description == "abnormal DNA methylation") %>%
group_by(gene, time) %>%
summarise(mean_expression = mean(log(expression))) %>%
arrange()
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 6 × 3
# Groups: gene [2]
gene time mean_expression
<chr> <dbl> <dbl>
1 Xist 0 6.95
2 Xist 4 6.34
3 Xist 8 7.13
4 Zdbf2 0 6.27
5 Zdbf2 4 6.27
6 Zdbf2 8 6.19Remodeler les données
Dans le tibble rna, les lignes contiennent des valeurs
d’expression (l’unité) qui sont associées à une combinaison de 2 autres
variables : gene et sample.
Toutes les autres colonnes correspondent à des variables décrivant soit l’échantillon (organisme, âge, sexe, …) soit le gène (gène_biotype, ENTREZ_ID, produit, …). Les variables qui ne changent pas avec les gènes ou avec les échantillons auront la même valeur dans toutes les lignes.
R
rna %>%
arrange(gene)
SORTIE
# A tibble: 32,428 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 AI504432 GSM25… 1230 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 AI504432 GSM25… 1085 Mus mus… 8 Fema… NonInfec… C57BL… 0 Cereb…
3 AI504432 GSM25… 969 Mus mus… 8 Fema… NonInfec… C57BL… 0 Cereb…
4 AI504432 GSM25… 1284 Mus mus… 8 Fema… Influenz… C57BL… 4 Cereb…
5 AI504432 GSM25… 966 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
6 AI504432 GSM25… 918 Mus mus… 8 Male Influenz… C57BL… 8 Cereb…
7 AI504432 GSM25… 985 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 AI504432 GSM25… 972 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
9 AI504432 GSM25… 1000 Mus mus… 8 Fema… Influenz… C57BL… 4 Cereb…
10 AI504432 GSM25… 816 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Cette structure est appelée « format long », car une colonne contient toutes les valeurs, et d’autres colonnes répertorient le contexte de la valeur.
Dans certains cas, le « format long » n’est pas vraiment « lisible par l’homme », et un autre format, un « format large » est préféré, comme manière plus compacte de représenter les données. This is typically the case with gene expression values that scientists are used to look as matrices, were rows represent genes and columns represent samples.
Dans ce format, il deviendrait donc simple d’explorer la relation entre les niveaux d’expression génique au sein et entre les échantillons.
SORTIE
# A tibble: 1,474 × 23
gene GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988
2 Apod 36194 10347 9173 10620 13021 29594
3 Cyp2d22 4060 1616 1603 1901 2171 3349
4 Klk6 287 629 641 578 448 195
5 Fcrls 85 233 244 237 180 38
6 Slc2a4 782 231 248 265 313 786
7 Exd2 1619 2288 2235 2513 2366 1359
8 Gjc2 288 595 568 551 310 146
9 Plp1 43217 101241 96534 58354 53126 27173
10 Gnb4 1071 1791 1867 1430 1355 798
# ℹ 1,464 more rows
# ℹ 16 more variables: GSM2545342 <dbl>, GSM2545343 <dbl>, GSM2545344 <dbl>,
# GSM2545345 <dbl>, GSM2545346 <dbl>, GSM2545347 <dbl>, GSM2545348 <dbl>,
# GSM2545349 <dbl>, GSM2545350 <dbl>, GSM2545351 <dbl>, GSM2545352 <dbl>,
# GSM2545353 <dbl>, GSM2545354 <dbl>, GSM2545362 <dbl>, GSM2545363 <dbl>,
# GSM2545380 <dbl>Pour convertir les valeurs d’expression génique de rna
en un format large, nous devons créer une nouvelle table où les valeurs
de la colonne sample deviendraient les noms des variables
de colonne.
The key point here is that we are still following a tidy data structure, but we have reshaped the data according to the observations of interest: expression levels per gene instead of recording them per gene and per sample.
La transformation inverse serait de transformer les noms de colonnes en valeurs d’une nouvelle variable.
Nous pouvons effectuer ces deux transformations avec deux fonctions
tidyr, pivot_longer() et
pivot_wider() (voir ici
pour détails).
Pivoter les données dans un format plus large
Sélectionnons les 3 premières colonnes de rna et
utilisons pivot_wider() pour transformer les données en
grand format.
R
rna_exp <- rna %>%
select(gene, sample, expression)
rna_exp
SORTIE
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Cyp2d22 GSM2545336 4060
4 Klk6 GSM2545336 287
5 Fcrls GSM2545336 85
6 Slc2a4 GSM2545336 782
7 Exd2 GSM2545336 1619
8 Gjc2 GSM2545336 288
9 Plp1 GSM2545336 43217
10 Gnb4 GSM2545336 1071
# ℹ 32,418 more rowspivot_wider prend trois arguments principaux :
- les données à transformer ;
- le
names_from: la colonne dont les valeurs deviendront de nouveaux noms de colonne ; - les
values_from: la colonne dont les valeurs rempliront les nouvelles colonnes .
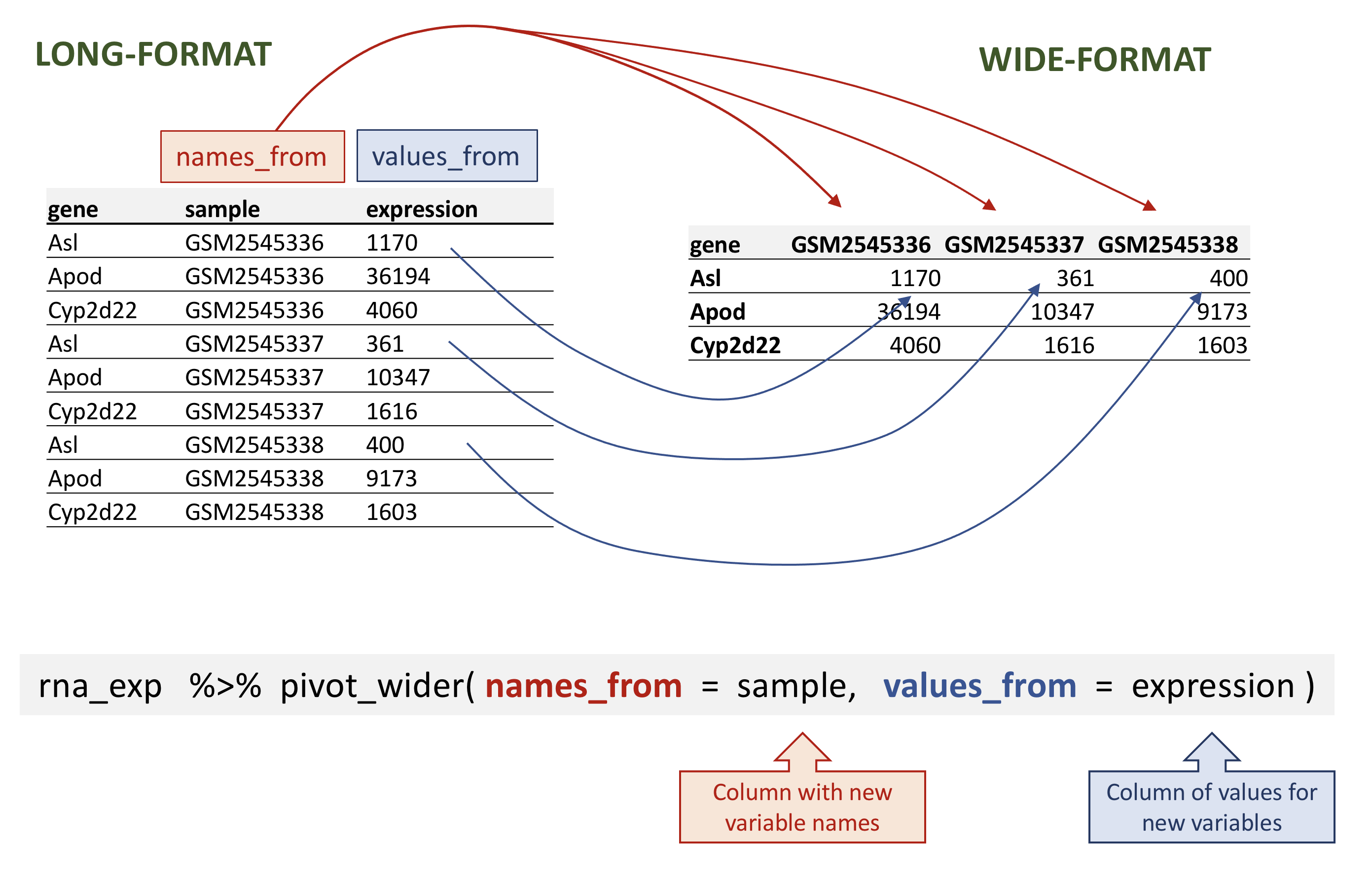
rna.
R
rna_wide <- rna_exp %>%
pivot_wider(names_from = sample,
values_from = expression)
rna_wide
SORTIE
# A tibble: 1,474 × 23
gene GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988
2 Apod 36194 10347 9173 10620 13021 29594
3 Cyp2d22 4060 1616 1603 1901 2171 3349
4 Klk6 287 629 641 578 448 195
5 Fcrls 85 233 244 237 180 38
6 Slc2a4 782 231 248 265 313 786
7 Exd2 1619 2288 2235 2513 2366 1359
8 Gjc2 288 595 568 551 310 146
9 Plp1 43217 101241 96534 58354 53126 27173
10 Gnb4 1071 1791 1867 1430 1355 798
# ℹ 1,464 more rows
# ℹ 16 more variables: GSM2545342 <dbl>, GSM2545343 <dbl>, GSM2545344 <dbl>,
# GSM2545345 <dbl>, GSM2545346 <dbl>, GSM2545347 <dbl>, GSM2545348 <dbl>,
# GSM2545349 <dbl>, GSM2545350 <dbl>, GSM2545351 <dbl>, GSM2545352 <dbl>,
# GSM2545353 <dbl>, GSM2545354 <dbl>, GSM2545362 <dbl>, GSM2545363 <dbl>,
# GSM2545380 <dbl>Notez que par défaut, la fonction pivot_wider() ajoutera
NA pour les valeurs manquantes.
Imaginons que, pour une raison quelconque, nous ayons des valeurs d’expression manquantes pour certains gènes dans certains échantillons. In the following fictive example, the gene Cyp2d22 has only one expression value, in GSM2545338 sample.
R
rna_with_missing_values <- rna %>%
select(gene, sample, expression) %>%
filter(gene %in% c("Asl", "Apod", "Cyp2d22")) %>%
filter(sample %in% c("GSM2545336", "GSM2545337", "GSM2545338")) %>%
arrange(sample) %>%
filter(!(gene == "Cyp2d22" & sample != "GSM2545338"))
rna_with_missing_values
SORTIE
# A tibble: 7 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Asl GSM2545337 361
4 Apod GSM2545337 10347
5 Asl GSM2545338 400
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545338 1603Par défaut, la fonction pivot_wider() ajoutera
NA pour les valeurs manquantes. Ceci peut être paramétré
avec l’argument values_fill de la fonction
pivot_wider().
R
rna_with_missing_values %>%
pivot_wider(names_from = échantillon,
values_from = expression)
ERREUR
Error in `pivot_wider()`:
! Can't select columns that don't exist.
✖ Column `échantillon` doesn't exist.R
rna_with_missing_values %>%
pivot_wider(names_from = échantillon,
values_from = expression,
valeurs_fill = 0)
ERREUR
Error in `pivot_wider()`:
! Can't select columns that don't exist.
✖ Column `échantillon` doesn't exist.Pivoter les données dans un format plus long
Dans la situation inverse, nous utilisons les noms de colonnes et les transformons en une paire de nouvelles variables. Une variable représente les noms de colonnes sous forme de valeurs , et l’autre variable contient les valeurs précédemment associées aux noms de colonnes.
pivot_longer() prend quatre arguments principaux :
- les données à transformer ;
- le
names_to: le nouveau nom de colonne que nous souhaitons créer et remplir avec les noms de colonnes actuels ; - les
values_to: le nouveau nom de colonne que nous souhaitons créer et remplir avec valeurs actuelles ; - les noms des colonnes à utiliser pour renseigner les variables
names_toetvalues_to(ou à supprimer).

rna.
To recreate rna_long from rna_wide we would
create a key called sample and value called
expression and use all columns except gene for
the key variable. Here we drop gene column with a minus
sign.
Notice how the new variable names are to be quoted here.
R
rna_long <- rna_wide %>%
pivot_longer(names_to = "sample",
values_to = "expression",
-gene)
rna_long
SORTIE
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Asl GSM2545339 586
5 Asl GSM2545340 626
6 Asl GSM2545341 988
7 Asl GSM2545342 836
8 Asl GSM2545343 535
9 Asl GSM2545344 586
10 Asl GSM2545345 597
# ℹ 32,418 more rowsNous aurions également pu utiliser une spécification indiquant les
colonnes à inclure. Cela peut être utile si vous disposez d’un grand
nombre de colonnes d’identification , et il est plus facile de spécifier
ce qu’il faut rassembler que ce qu’il faut laisser seul. Ici, la
fonction starts_with() peut aider à récupérer des exemples
de noms sans avoir à tous les lister ! Une autre possibilité serait
d’utiliser l’opérateur : !
R
rna_wide %>%
pivot_longer(names_to = "sample",
values_to = "expression",
cols = start_with("GSM"))
ERREUR
Error in `pivot_longer()`:
ℹ In argument: `start_with("GSM")`.
Caused by error in `start_with()`:
! could not find function "start_with"R
rna_wide %> %
pivot_longer(names_to = "échantillon",
valeurs_to = "expression",
GSM2545336:GSM2545380)
ERREUR
Error in rna_wide %> % pivot_longer(names_to = "échantillon", valeurs_to = "expression", : could not find function "%> %"Notez que si nous avions des valeurs manquantes dans le format large, le « NA » serait inclus dans le nouveau format long.
Souvenez-vous de notre précédent tibble fictif contenant des valeurs manquantes :
R
rna_with_missing_values
SORTIE
# A tibble: 7 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Asl GSM2545337 361
4 Apod GSM2545337 10347
5 Asl GSM2545338 400
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545338 1603R
wide_with_NA <- rna_with_missing_values %>%
pivot_wider(names_from = sample,
values_from = expression)
wide_with_NA
SORTIE
# A tibble: 3 × 4
gene GSM2545336 GSM2545337 GSM2545338
<chr> <dbl> <dbl> <dbl>
1 Asl 1170 361 400
2 Apod 36194 10347 9173
3 Cyp2d22 NA NA 1603R
wide_with_NA %>%
pivot_longer(names_to = "sample",
values_to = "expression",
-gene)
SORTIE
# A tibble: 9 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Apod GSM2545336 36194
5 Apod GSM2545337 10347
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545336 NA
8 Cyp2d22 GSM2545337 NA
9 Cyp2d22 GSM2545338 1603Passer à des formats plus larges et plus longs peut être un moyen utile d’équilibrer un ensemble de données afin que chaque réplique ait la même composition.
R
rna1 <- rna %>%
select(gène, souris, expression) %>%
pivot_wider(names_from = souris, valeurs_from = expression)
ERREUR
Error in `select()`:
! Can't select columns that don't exist.
✖ Column `gène` doesn't exist.R
rna1
ERREUR
Error in eval(expr, envir, enclos): object 'rna1' not foundR
rna1 %>%
pivot_longer(names_to = "mouse_id", valeurs_to = "counts", -gene)
ERREUR
Error in eval(expr, envir, enclos): object 'rna1' not foundERREUR
Error in `pivot_longer()`:
! Arguments in `...` must be used.
✖ Problematic argument:
• valeurs_to = "counts"
ℹ Did you misspell an argument name?Question
Sous-ensemble de gènes situés sur les chromosomes X et Y de la trame de données « rna » et répartissent la trame de données avec « sexe » en colonnes, « nom_chromosome » en lignes et l’expression moyenne des gènes localisés dans chaque chromosome comme valeurs, comme dans le tableau suivant :
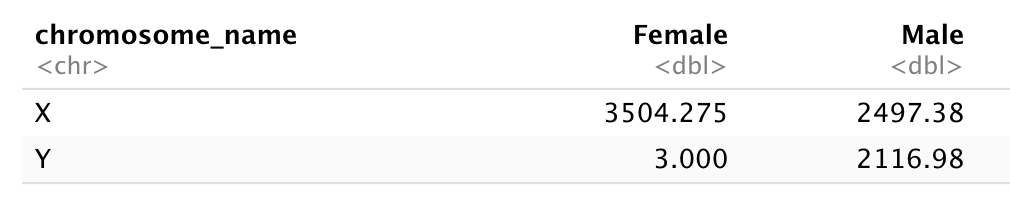
Il faudra résumer avant de remodeler !
Calculons d’abord le niveau d’expression moyen des gènes liés X et Y à partir de échantillons mâles et femelles…
R
arn %>%
filter(chromosome_name == "Y" | chromosome_name == "X") %>%
group_by(sex, chromosome_name) %>%
résumé(moyenne = moyenne(expression))
ERREUR
Error in résumé(., moyenne = moyenne(expression)): could not find function "résumé"Et faites pivoter le tableau au format large
R
rna_1 <- rna %>%
filter(chromosome_name == "Y" | chromosome_name == "X") %>%
group_by(sex, chromosome_name) %>%
summarise(mean = moyenne(expression)) %>%
pivot_wider(names_from = sexe,
valeurs_from = moyenne)
ERREUR
Error in `summarise()`:
ℹ In argument: `mean = moyenne(expression)`.
ℹ In group 1: `sex = "Female"` and `chromosome_name = "X"`.
Caused by error in `moyenne()`:
! could not find function "moyenne"R
rna_1
ERREUR
Error in eval(expr, envir, enclos): object 'rna_1' not foundMaintenant, prenez cette trame de données et transformez-la avec
pivot_longer() afin que chaque ligne soit une combinaison
unique de chromosome_name par gender.
R
rna_1 %>%
pivot_longer(names_to = "gender",
valeurs_to = "mean",
-chromosome_name)
ERREUR
Error in eval(expr, envir, enclos): object 'rna_1' not foundERREUR
Error in `pivot_longer()`:
! Arguments in `...` must be used.
✖ Problematic argument:
• valeurs_to = "mean"
ℹ Did you misspell an argument name?Calculons d’abord l’expression moyenne par gène et par temps
R
arn %>%
group_by(gène, temps) %>%
résumé(mean_exp = moyenne(expression))
ERREUR
Error in résumé(., mean_exp = moyenne(expression)): could not find function "résumé"avant d’utiliser la fonction pivot_wider()
R
rna_time <- rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.R
rna_time
SORTIE
# A tibble: 1,474 × 4
# Groups: gene [1,474]
gene `0` `4` `8`
<chr> <dbl> <dbl> <dbl>
1 AI504432 1034. 1104. 1014
2 AW046200 155. 152. 81
3 AW551984 238 302. 342.
4 Aamp 4603. 4870 4763.
5 Abca12 5.29 4.25 4.14
6 Abcc8 2576. 2609. 2292.
7 Abhd14a 591. 547. 432.
8 Abi2 4881. 4903. 4945.
9 Abi3bp 1175. 1061. 762.
10 Abl2 2170. 2078. 2131.
# ℹ 1,464 more rowsNotez que cela génère un tibble avec certains noms de colonnes commençant par un nombre. Si nous voulions sélectionner la colonne correspondant aux points temporels, nous ne pourrions pas utiliser directement les noms de colonnes… Que se passe-t-il lorsque l’on sélectionne la colonne 4 ?
R
rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
select(gene, 4)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene `8`
<chr> <dbl>
1 AI504432 1014
2 AW046200 81
3 AW551984 342.
4 Aamp 4763.
5 Abca12 4.14
6 Abcc8 2292.
7 Abhd14a 432.
8 Abi2 4945.
9 Abi3bp 762.
10 Abl2 2131.
# ℹ 1,464 more rowsPour sélectionner le timepoint 4, il faudrait citer le nom de la colonne, avec des backticks “\`”
R
rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
select(gene, `4`)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene `4`
<chr> <dbl>
1 AI504432 1104.
2 AW046200 152.
3 AW551984 302.
4 Aamp 4870
5 Abca12 4.25
6 Abcc8 2609.
7 Abhd14a 547.
8 Abi2 4903.
9 Abi3bp 1061.
10 Abl2 2078.
# ℹ 1,464 more rowsUne autre possibilité serait de renommer la colonne, en choisissant un nom qui ne commence pas par un chiffre :
R
rna %>%
group_by(gene, time) %>%
summarise(mean_exp = mean(expression)) %>%
pivot_wider(names_from = time,
values_from = mean_exp) %>%
rename("time0" = `0`, "time4" = `4`, "time8" = `8`) %>%
select(gene, time4)
SORTIE
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.SORTIE
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene time4
<chr> <dbl>
1 AI504432 1104.
2 AW046200 152.
3 AW551984 302.
4 Aamp 4870
5 Abca12 4.25
6 Abcc8 2609.
7 Abhd14a 547.
8 Abi2 4903.
9 Abi3bp 1061.
10 Abl2 2078.
# ℹ 1,464 more rowsQuestion
Utilisez la trame de données précédente contenant les niveaux d’expression moyens par point temporel et créez une nouvelle colonne contenant les changements de pli entre le point temporel 8 et le point temporel 0, et les changements de pli entre le point temporel 8 et le point temporel 4. Convertissez ce tableau en un tableau au format long regroupant les changements de pli calculés.
À partir du tibble rna_time :
R
arn_time
ERREUR
Error in eval(expr, envir, enclos): object 'arn_time' not foundCalculer les changements de plis :
R
rna_time %>%
muter (time_8_vs_0 = `8` / `0`, time_8_vs_4 = `8` / `4`)
ERREUR
Error in muter(., time_8_vs_0 = `8`/`0`, time_8_vs_4 = `8`/`4`): could not find function "muter"Et utilisez la fonction pivot_longer() :
R
rna_time %>%
mutate(time_8_vs_0 = `8` / `0`, time_8_vs_4 = `8` / `4`) %>%
pivot_longer(names_to = "comparisons",
values_to = "Fold_changes",
time_8_vs_0:time_8_vs_4)
SORTIE
# A tibble: 2,948 × 6
# Groups: gene [1,474]
gene `0` `4` `8` comparisons Fold_changes
<chr> <dbl> <dbl> <dbl> <chr> <dbl>
1 AI504432 1034. 1104. 1014 time_8_vs_0 0.981
2 AI504432 1034. 1104. 1014 time_8_vs_4 0.918
3 AW046200 155. 152. 81 time_8_vs_0 0.522
4 AW046200 155. 152. 81 time_8_vs_4 0.532
5 AW551984 238 302. 342. time_8_vs_0 1.44
6 AW551984 238 302. 342. time_8_vs_4 1.13
7 Aamp 4603. 4870 4763. time_8_vs_0 1.03
8 Aamp 4603. 4870 4763. time_8_vs_4 0.978
9 Abca12 5.29 4.25 4.14 time_8_vs_0 0.784
10 Abca12 5.29 4.25 4.14 time_8_vs_4 0.975
# ℹ 2,938 more rowsJoindre des tables
Dans de nombreuses situations réelles, les données sont réparties sur plusieurs tables. Cela se produit généralement parce que différents types d’informations sont collectés à partir de différentes sources.
Il peut être souhaitable que certaines analyses combinent les données de deux ou plusieurs tables en une seule trame de données basée sur une colonne qui serait commune à toutes les tables.
Le package dplyr fournit un ensemble de fonctions de
jointure pour combiner deux trames de données basées sur des
correspondances dans des colonnes spécifiées. Ici, nous fournissons une
brève introduction aux jointures. Pour en savoir plus, veuillez vous
référer au chapitre sur les jointures de table . La Data Transformation
Cheat Sheet fournit également un bref aperçu sur les jointures de
table.
Nous allons illustrer la jointure en utilisant une petite table,
rna_mini que nous allons créer en sous-définissant la table
rna d’origine, en ne gardant que 3 colonnes et 10
lignes.
R
rna_mini <- rna %>%
select(gène, échantillon, expression) %>%
head(10)
ERREUR
Error in `select()`:
! Can't select columns that don't exist.
✖ Column `gène` doesn't exist.R
rna_mini
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundLe deuxième tableau, annot1, contient 2 colonnes, gene
et gene_description. Vous pouvez soit télécharger
annot1.csv en cliquant sur le lien puis en vous déplaçant dans le
dossier data/, ou vous pouvez utiliser le code R ci-dessous
pour le télécharger directement dans le dossier.
R
download.file(url = "https://carpentries-incubator.github.io/bioc-intro/data/annot1.csv",
destfile = "data/annot1.csv")
annot1 <- read_csv(file = "data/annot1.csv")
annot1
SORTIE
# A tibble: 10 × 2
gene gene_description
<chr> <chr>
1 Cyp2d22 cytochrome P450, family 2, subfamily d, polypeptide 22 [Source:MGI S…
2 Klk6 kallikrein related-peptidase 6 [Source:MGI Symbol;Acc:MGI:1343166]
3 Fcrls Fc receptor-like S, scavenger receptor [Source:MGI Symbol;Acc:MGI:19…
4 Plp1 proteolipid protein (myelin) 1 [Source:MGI Symbol;Acc:MGI:97623]
5 Exd2 exonuclease 3'-5' domain containing 2 [Source:MGI Symbol;Acc:MGI:192…
6 Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88056]
7 Gnb4 guanine nucleotide binding protein (G protein), beta 4 [Source:MGI S…
8 Slc2a4 solute carrier family 2 (facilitated glucose transporter), member 4 …
9 Asl argininosuccinate lyase [Source:MGI Symbol;Acc:MGI:88084]
10 Gjc2 gap junction protein, gamma 2 [Source:MGI Symbol;Acc:MGI:2153060] Nous voulons maintenant joindre ces deux tables en une seule
contenant toutes les variables en utilisant la fonction
full_join() du package dplyr. La fonction
trouvera automatiquement la variable commune correspondant aux colonnes
de la première et de la deuxième table. Dans ce cas, « gène » est la
variable commune . Ces variables sont appelées clés. Les clés sont
utilisées pour faire correspondre observations dans différentes
tables.
R
full_join(rna_mini, annot1)
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundDans la vraie vie, les annotations génétiques sont parfois étiquetées différemment.
La table annot2 est exactement la même que
annot1 sauf que la variable contenant les noms de gènes est
étiquetée différemment. Encore une fois, soit télécharger
annot2.csv vous-même et déplacez-le vers data/ ou
utilisez le code R ci-dessous.
R
download.file(url = "https://carpentries-incubator.github.io/bioc-intro/data/annot2.csv",
destfile = "data/annot2.csv")
annot2 <- read_csv(file = "data/annot2.csv")
annot2
SORTIE
# A tibble: 10 × 2
external_gene_name description
<chr> <chr>
1 Cyp2d22 cytochrome P450, family 2, subfamily d, polypeptide 22 [S…
2 Klk6 kallikrein related-peptidase 6 [Source:MGI Symbol;Acc:MGI…
3 Fcrls Fc receptor-like S, scavenger receptor [Source:MGI Symbol…
4 Plp1 proteolipid protein (myelin) 1 [Source:MGI Symbol;Acc:MGI…
5 Exd2 exonuclease 3'-5' domain containing 2 [Source:MGI Symbol;…
6 Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88056]
7 Gnb4 guanine nucleotide binding protein (G protein), beta 4 [S…
8 Slc2a4 solute carrier family 2 (facilitated glucose transporter)…
9 Asl argininosuccinate lyase [Source:MGI Symbol;Acc:MGI:88084]
10 Gjc2 gap junction protein, gamma 2 [Source:MGI Symbol;Acc:MGI:…Si aucun des noms de variables ne correspond, nous pouvons définir
manuellement les variables à utiliser pour la correspondance. Ces
variables peuvent être définies en utilisant l’argument by,
comme indiqué ci-dessous avec les tables rna_mini et
annot2.
R
full_join(rna_mini, annot2, by = c("gene" = "external_gene_name"))
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundComme on peut le voir ci-dessus, le nom de variable de la première table est conservé dans celle jointe.
Défi:
Téléchargez la table annot3 en cliquant sur ici
et placez la table dans votre Dépôt de données. À l’aide de la fonction
full_join() , joignez les tables rna_mini et
annot3. Que s’est-il passé pour les gènes Klk6,
mt-Tf, mt-Rnr1, mt-Tv, mt-Rnr2 et
mt-Tl1 ?
R
annot3 <- read_csv("data/annot3.csv")
full_join(rna_mini, annot3)
ERREUR
Error in eval(expr, envir, enclos): object 'rna_mini' not foundLes gènes Klk6 ne sont présents que dans
rna_mini, tandis que les gènes mt-Tf,
mt-Rnr1, mt-Tv, mt-Rnr2 et mt-Tl1
sont présent uniquement dans la table annot3. Leurs valeurs
respectives pour les variables du tableau ont été codées comme
manquantes.
Exporter des données
Maintenant que vous avez appris à utiliser dplyr pour
extraire des informations de ou résumer vos données brutes, vous
souhaiterez peut-être exporter ces nouveaux ensembles de données pour
les partager avec vos collaborateurs ou pour les archiver.
Semblable à la fonction read_csv() utilisée pour lire
les fichiers CSV dans R, il existe une fonction write_csv()
qui génère des fichiers CSV à partir de trames de données.
Avant d’utiliser write_csv(), nous allons créer un
nouveau dossier, data_output, dans notre répertoire de
travail qui stockera cet ensemble de données généré. Nous ne voulons pas
que écrive les ensembles de données générés dans le même répertoire que
nos données brutes. C’est une bonne pratique de les garder séparés. Le
dossier data ne doit contenir que les données brutes et non
modifiées, et doit être laissé seul pour nous assurer que nous ne
supprimons pas ou ne le modifions pas. En revanche, notre script
générera le contenu du répertoire data_output , donc même
si les fichiers qu’il contient sont supprimés, nous pouvons toujours les
régénérer.
Utilisons write_csv() pour sauvegarder la table rna_wide
que nous avons créée précédemment.
R
write_csv(rna_wide, file = "data_output/rna_wide.csv")
Content from Visualisation de données
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 120 minutes
Vue d'ensemble
Questions
- Visualisation en R
Objectifs
- Produisez des nuages de points, des boxplots, des tracés linéaires, etc. en utilisant ggplot.
- Définissez les paramètres de tracé universels.
- Décrivez ce qu’est le facettage et appliquez le facettage dans ggplot.
- Modifiez l’esthétique d’un tracé ggplot existant (y compris les étiquettes des axes et la couleur).
- Créez des tracés complexes et personnalisés à partir de données dans un bloc de données.
Cet épisode est basé sur la leçon Analyse des données et Visualisation dans R pour les écologistes de Data Carpentries.
Visualisation de données
Nous commençons par charger les packages requis.
ggplot2 est inclus dans le package
tidyverse.
R
bibliothèque("tidyverse")
ERREUR
Error in bibliothèque("tidyverse"): could not find function "bibliothèque"Si vous n’êtes pas encore dans l’espace de travail, chargez les données que nous avons enregistrées dans la leçon précédente.
R
arn <- read.csv("data/rnaseq.csv")
La Data Visualization Cheat
couvrira les bases et les fonctionnalités plus avancées de
ggplot2 et aidera, en plus de servir de rappel, à obtenir
un aperçu des nombreuses représentations de données disponibles dans le
package. Les didacticiels vidéo suivants (partie 1 et 2) de Thomas
Lin Pedersen sont également très instructifs.
Tracer avec ggplot2
ggplot2 est un package de traçage qui simplifie la
création de tracés complexes à partir de données dans un bloc de
données. Il fournit une interface plus programmatique pour spécifier les
variables à tracer, comment elles sont affichées, et les propriétés
visuelles générales. La fondation théorique qui prend en charge le
ggplot2 est la Grammar of Graphics (@Wilkinson :2005). En utilisant cette approche ,
nous n’avons besoin que de changements minimes si les données
sous-jacentes changent ou si nous décidons de passer d’un diagramme à
barres à un nuage de points. Cela aide à créer des tracés de qualité de
publication avec un minimum d’ajustements et de peaufinages.
Il existe un livre sur ggplot2 (@ggplot2book) qui fournit un bon aperçu de ,
mais il est obsolète. La 3ème édition est en préparation et sera disponible gratuitement en ligne.
La page Web ggplot2 (https://ggplot2.tidyverse.org)
fournit une documentation abondante.
ggplot2 fonctionne comme des données au format « long »,
c’est-à-dire une colonne pour chaque dimension et une ligne pour chaque
observation. Des données bien structurées vous feront gagner beaucoup de
temps lors de la création de chiffres avec ggplot2.
Les graphiques ggplot sont construits étape par étape en ajoutant de nouveaux éléments. L’ajout de couches de cette manière permet une grande flexibilité et une personnalisation des tracés.
L’idée derrière la grammaire graphique est que vous pouvez construire chaque graphique à partir des 3 mêmes composants : (1) un ensemble de données, (2) un système de coordonnées, et (3) des géoms. — c’est-à-dire des marques visuelles qui représentent des points de données [^trois\_comp\_ggplot2]
Pour construire un ggplot, nous utiliserons le modèle de base suivant qui peut être utilisé pour différents types de tracés :
ggplot(data = <DATA>, mapping = aes(<MAPPINGS>)) + <GEOM_FUNCTION>()- utilisez la fonction
ggplot()et liez le tracé à un cadre **data ** spécifique en utilisant l’argumentdata
R
ggplot(data = rna)
- définir un mapping (en utilisant la fonction
esthétique (
aes)), en sélectionnant les variables à tracer et en spécifiant comment les présenter dans le graphique, par exemple sous la forme x/ y positions ou caractéristiques telles que taille, forme, couleur, etc.
R
ggplot(data = rna, mapping = aes(x = expression))
-
ajoutez ‘geoms’ - géométries ou représentations graphiques des données dans le tracé (points, lignes, barres).
ggplot2propose de nombreuses géométries différentes ; nous en utiliserons quelques-uns courants aujourd’hui, notamment :* `geom_point()` pour les nuages de points, les diagrammes de points, etc. * `geom_histogram()` pour les histogrammes * `geom_boxplot()` pour, eh bien, les boxplots ! * `geom_line()` pour les lignes de tendance, les séries chronologiques, etc.
Pour ajouter une géométrie au tracé, utilisez l’opérateur
+. Utilisons d’abord geom_histogram() :
R
ggplot(data = rna, mapping = aes(x = expression)) +
geom_histogram()
ERREUR
Error in ggplot(data = rna, mapping = aes(x = expression)): could not find function "ggplot"Le + dans le package ggplot2 est
particulièrement utile car il vous permet de modifier les objets
ggplot existants. Cela signifie que vous pouvez facilement
configurer des modèles de tracé et explorer facilement différents types
de tracés, de sorte que le tracé ci-dessus peut également être généré
avec un code comme celui-ci :
R
# Assign plot to a variable
rna_plot <- ggplot(data = rna,
mapping = aes(x = expression))
# Draw the plot
rna_plot + geom_histogram()
R
# change bins
ggplot(rna, aes(x = expression)) +
geom_histogram(bins = 15)
ERREUR
Error in ggplot(rna, aes(x = expression)): could not find function "ggplot"R
# change binwidth
ggplot(rna, aes(x = expression)) +
geom_histogram(binwidth = 2000)
ERREUR
Error in ggplot(rna, aes(x = expression)): could not find function "ggplot"Nous pouvons observer ici que les données sont biaisées vers la
droite. Nous pouvons appliquer la transformation log2 pour avoir une
distribution plus symétrique. Notez que nous ajoutons ici une petite
valeur constante (+1) pour éviter que les valeurs
-Inf soient renvoyées pour les valeurs d’expression égales
à 0.
R
arn <- arn %>%
muter(expression_log = log2(expression + 1))
ERREUR
Error in arn %>% muter(expression_log = log2(expression + 1)): could not find function "%>%"Si l’on dessine maintenant l’histogramme des expressions transformées en log2, la distribution est en effet plus proche d’une distribution normale.
R
ggplot(rna, aes(x = expression_log)) + geom_histogram()
ERREUR
Error in ggplot(rna, aes(x = expression_log)): could not find function "ggplot"À partir de maintenant, nous travaillerons sur les valeurs d’expression transformées en log.
Défi
Une autre façon de visualiser cette transformation est de considérer l’échelle des observations. Par exemple, il peut être intéressant de changer l’échelle de l’axe pour mieux répartir les observations dans l’espace de la parcelle . Changer l’échelle des axes se fait de la même manière que ajouter/modifier d’autres composants (c’est-à-dire en ajoutant progressivement des commandes ). Essayez de faire cette modification :
- Représenter l’expression non transformée sur l’échelle log10 ; voir
scale_x_log10(). Comparez-le avec le graphique précédent. Pourquoi des messages d’avertissement apparaissent-ils maintenant ?
R
ggplot(data = rna,mapping = aes(x = expression))+
geom_histogram() +
scale_x_log10()
ERREUR
Error in ggplot(data = rna, mapping = aes(x = expression)): could not find function "ggplot"Remarques
- Tout ce que vous mettez dans la fonction
ggplot()peut être vu par n’importe quelle couche geom que vous ajoutez (c’est-à-dire qu’il s’agit de paramètres de tracé globaux). Ce inclut le mappage des axes x et y que vous avez configuré dansaes(). - Vous pouvez également spécifier des mappages pour une géom donnée
indépendamment des mappages définis globalement dans la fonction
ggplot(). - Le signe
+utilisé pour ajouter de nouveaux calques doit être placé à la fin de la ligne contenant le calque précédent. If, instead, the+sign is added at the beginning of the line containing the new layer,ggplot2will not add the new layer and will return an error message.
R
# This is the correct syntax for adding layers
rna_plot +
geom_histogram()
# This will not add the new layer and will return an error message
rna_plot
+ geom_histogram()
Construire vos parcelles de manière itérative
Nous allons maintenant dessiner un nuage de points avec deux
variables continues et la fonction geom_point(). Ce
graphique représentera les changements de pli log2 de l’expression
comparant le temps 8 au temps 0 et le temps 4 au temps 0. À cette fin,
nous devons d’abord calculer les moyennes des valeurs d’expression
transformées en log par gène et par temps, puis le pli du log change en
en soustrayant les expressions du log moyen entre le temps 8 et le temps
0. et entre le temps 4 et le temps 0. Notez que nous incluons également
ici le biotype du gène que nous utiliserons plus tard pour représenter
les gènes. Nous enregistrerons les changements de repli dans un nouveau
bloc de données appelé rna_fc.
R
rna_fc <- rna %>% select(gene, time,
gene_biotype, expression_log) %>%
group_by(gene, time, gene_biotype) %>%
summary(mean_exp = moyenne (expression_log)) %>%
pivot_wider(names_from = temps,
valeurs_from = moyenne_exp) %>%
mutate(time_8_vs_0 = `8` - `0`, time_4_vs_0 = `4` - `0`)
ERREUR
Error in rna %>% select(gene, time, gene_biotype, expression_log) %>% : could not find function "%>%"Nous pouvons ensuite construire un ggplot avec l’ensemble de données
nouvellement créé rna_fc. Construire des parcelles avec
ggplot2 est généralement un processus itératif. Nous
commençons par définir l’ensemble de données que nous allons utiliser,
tracer les axes et choisir une géom :
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point()
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)): could not find function "ggplot"Ensuite, nous commençons à modifier ce tracé pour en extraire plus d’informations. Par exemple, nous pouvons ajouter de la transparence (« alpha ») pour éviter le surtraçage :
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3)
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)): could not find function "ggplot"On peut également ajouter des couleurs pour tous les points :
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3, color = "blue")
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)): could not find function "ggplot"Ou pour colorer différemment chaque gène du tracé, vous pouvez
utiliser un vecteur comme entrée dans l’argument color.
ggplot2 fournira une couleur différente correspondant à
différentes valeurs dans le vecteur. Voici un exemple où nous colorons
avec gene_biotype :
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3, aes(color = gene_biotype))
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)): could not find function "ggplot"Nous pouvons également spécifier les couleurs directement à
l’intérieur du mappage fourni dans la fonction ggplot().
Cela sera visible par toutes les couches géométriques et la cartographie
sera déterminée par les axes x et y définis dans aes().
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_point(alpha = 0.3)
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0, : could not find function "ggplot"Enfin, nous pourrions également ajouter une ligne diagonale avec la
fonction geom_abline() :
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_point(alpha = 0.3) +
geom_abline(intercept = 0)
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0, : could not find function "ggplot"Notez que nous pouvons changer la couche géométrique de
geom_point à geom_jitter et les couleurs
seront toujours déterminées par gene_biotype.
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_jitter(alpha = 0.3) +
geom_abline(intercept = 0)
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0, : could not find function "ggplot"ERREUR
Error in bibliothèque("hexbin"): could not find function "bibliothèque"Défi
Les nuages de points peuvent être des outils d’exploration utiles
pour de petits ensembles de données. Pour les ensembles de données avec
un grand nombre d’observations, tels que l’ensemble de données
rna_fc , le surtraçage des points peut constituer une
limitation des nuages de points. Une stratégie pour gérer de tels
paramètres consiste à utiliser le regroupement hexagonal d’observations
. L’espace de l’intrigue est divisé en hexagones. Chaque hexagone se
voit attribuer une couleur en fonction du nombre d’observations qui
tombent dans ses limites.
Pour utiliser le regroupement hexagonal dans
ggplot2, installez d’abord le package Rhexbindepuis CRAN et chargez-le.Utilisez ensuite la fonction
geom_hex()pour produire la figure hexbin.Quelles sont les forces et les faiblesses relatives d’un diagramme hexagonal par rapport à un nuage de points ? Examinez le nuage de points ci-dessus et comparez-le avec le diagramme hexagonal que vous avez créé.
R
install.packages("hexbin")
R
library("hexbin")
ERREUR
Error in library("hexbin"): there is no package called 'hexbin'R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_hex() +
geom_abline(intercept = 0)
ERREUR
Error in ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)): could not find function "ggplot"R
ggplot(data = rna, mapping = aes(y = expression_log, x = sample)) +
geom_point(aes(color = time))
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"Boîte à moustaches
Nous pouvons utiliser des boxplots pour visualiser la distribution des expressions géniques au sein de chaque échantillon :
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_boxplot()
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"En ajoutant des points au boxplot, on peut avoir une meilleure idée du nombre de mesures et de leur répartition :
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_jitter(alpha = 0.2, color = "tomato") +
geom_boxplot(alpha = 0)
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"Nous devrions inverser l’ordre de ces deux géométries :
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_boxplot(alpha = 0) +
geom_jitter(alpha = 0.2, color = "tomato")
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"Vous remarquerez peut-être que les valeurs sur l’axe des x ne sont toujours pas correctement lisibles. Modifions l’orientation des étiquettes et ajustons-les verticalement et horizontalement afin qu’elles ne se chevauchent pas. Vous pouvez utiliser un angle de 90 degrés ou expérimenter pour trouver l’angle approprié pour les étiquettes orientées en diagonale :
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_jitter(alpha = 0.2, color = "tomato") +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"Défi
Ajoutez de la couleur aux points de données sur votre boxplot en fonction de la durée de l’infection (« time »).
Indice : Vérifiez la classe pour « heure ». Envisagez de
changer la classe de time d’entier pour prendre en compte
directement dans le mappage ggplot. Pourquoi cela change-t-il la façon
dont R crée le graphique ?
R
# time as integer
ggplot(data = rna,
mapping = aes(y = expression_log,
x = sample)) +
geom_jitter(alpha = 0.2, aes(color = time)) +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"R
# time as factor
ggplot(data = rna,
mapping = aes(y = expression_log,
x = sample)) +
geom_jitter(alpha = 0.2, aes(color = as.factor(time))) +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"Défi
Les boxplots sont des résumés utiles, mais cachent la forme de la distribution . Par exemple, si la distribution est bimodale, nous ne la verrions pas dans un boxplot. Une alternative au boxplot est le tracé en violon , où la forme (de la densité de points) est dessinée.
- Remplacez la boîte à moustaches par une intrigue en violon ; voir
geom_violin(). Remplissez les violons en fonction du temps avec l’argumentfill.
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_violin(aes(fill = as.factor(time))) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_violin(aes(fill = sex)) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
ERREUR
Error in ggplot(data = rna, mapping = aes(y = expression_log, x = sample)): could not find function "ggplot"Tracés linéaires
Calculons l’expression moyenne par durée de l’infection pour les 10
gènes ayant les changements logarithmiques les plus élevés en comparant
le temps 8 au temps 0. Tout d’abord, nous devons sélectionner les gènes
et créer un sous-ensemble de rna appelé
sub_rna contenant les 10 gènes sélectionnés, puis nous
devons regrouper les données et calculer l’expression moyenne des gènes
dans chaque groupe:
R
rna_fc <- rna_fc %>% arrange(desc(time_8_vs_0))
ERREUR
Error in rna_fc %>% arrange(desc(time_8_vs_0)): could not find function "%>%"R
genes_selected <- rna_fc$gene[1:10]
ERREUR
Error in eval(expr, envir, enclos): object 'rna_fc' not foundR
sub_rna <- rna %>%
filter(gene %in% genes_selected)
ERREUR
Error in rna %>% filter(gene %in% genes_selected): could not find function "%>%"R
mean_exp_by_time <- sub_rna %>%
group_by(gene,time) %>%
summarize(mean_exp = mean(expression_log))
ERREUR
Error in sub_rna %>% group_by(gene, time) %>% summarize(mean_exp = mean(expression_log)): could not find function "%>%"R
mean_exp_by_time
ERREUR
Error in eval(expr, envir, enclos): object 'mean_exp_by_time' not foundNous pouvons construire le tracé linéaire avec la durée de l’infection sur l’axe des x et l’expression moyenne sur l’axe des y :
R
ggplot(data = moyenne_exp_by_time, mapping = aes(x = temps, y = moyenne_exp)) +
geom_line()
ERREUR
Error in ggplot(data = moyenne_exp_by_time, mapping = aes(x = temps, y = moyenne_exp)): could not find function "ggplot"Malheureusement, cela ne fonctionne pas car nous avons tracé ensemble
les données de tous les gènes . Nous devons dire à ggplot de tracer une
ligne pour chaque gène en modifiant la fonction esthétique pour inclure
group = gene :
R
ggplot(data = moyenne_exp_by_time,
mapping = aes(x = temps, y = moyenne_exp, groupe = gène)) +
geom_line()
ERREUR
Error in ggplot(data = moyenne_exp_by_time, mapping = aes(x = temps, y = moyenne_exp, : could not find function "ggplot"Nous pourrons distinguer les gènes dans l’intrigue si nous ajoutons
des couleurs (l’utilisation de color regroupe également
automatiquement les données) :
R
ggplot(data = moyenne_exp_by_time,
mapping = aes(x = temps, y = moyenne_exp, couleur = gène)) +
geom_line()
ERREUR
Error in ggplot(data = moyenne_exp_by_time, mapping = aes(x = temps, y = moyenne_exp, : could not find function "ggplot"Facettage
ggplot2 a une technique spéciale appelée
faceting qui permet à l’utilisateur de diviser une parcelle en
plusieurs (sous) parcelles en fonction d’un facteur inclus dans
l’ensemble de données. Ces différentes sous-parcelles héritent des mêmes
propriétés (limites des axes, ticks, …) pour faciliter leur comparaison
directe. Nous allons l’utiliser pour créer un tracé linéaire dans le
temps pour chaque gène :
R
ggplot(data = moyenne_exp_by_time,
mapping = aes(x = temps, y = moyenne_exp)) + geom_line() +
facet_wrap(~ gène)
ERREUR
Error in ggplot(data = moyenne_exp_by_time, mapping = aes(x = temps, y = moyenne_exp)): could not find function "ggplot"Ici, les axes x et y ont la même échelle pour toutes les
sous-parcelles. Vous pouvez changer ce comportement par défaut en
modifiant scales afin d’autoriser une échelle libre pour
l’axe y :
R
ggplot(data = moyenne_exp_by_time,
mapping = aes(x = temps, y = moyenne_exp)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y")
ERREUR
Error in ggplot(data = moyenne_exp_by_time, mapping = aes(x = temps, y = moyenne_exp)): could not find function "ggplot"Nous aimerions maintenant diviser la ligne dans chaque parcelle selon le sexe des souris. Pour ce faire, nous devons calculer l’expression moyenne dans le bloc de données regroupé par « gène », « temps » et « sexe » :
R
Mean_exp_by_time_sex <- sub_rna %>%
group_by(gene, time, sex) %>%
summary(mean_exp = Mean(expression_log))
ERREUR
Error in sub_rna %>% group_by(gene, time, sex) %>% summary(mean_exp = Mean(expression_log)): could not find function "%>%"R
Mean_exp_by_time_sex
ERREUR
Error in eval(expr, envir, enclos): object 'Mean_exp_by_time_sex' not foundNous pouvons maintenant créer le tracé à facettes en le divisant
davantage par sexe en utilisant color (au sein d’un seul
tracé) :
R
ggplot(data = moyenne_exp_by_time_sex,
mapping = aes(x = temps, y = moyenne_exp, couleur = sexe)) +
geom_line() +
facet_wrap(~ gène, échelles = "free_y")
ERREUR
Error in ggplot(data = moyenne_exp_by_time_sex, mapping = aes(x = temps, : could not find function "ggplot"Généralement, les tracés sur fond blanc semblent plus lisibles une
fois imprimés. Nous pouvons définir l’arrière-plan en blanc en utilisant
la fonction theme_bw(). De plus, nous pouvons supprimer la
grille :
R
ggplot(data = moyenne_exp_by_time_sex,
mapping = aes(x = temps, y = moyenne_exp, couleur = sexe)) +
geom_line() +
facet_wrap(~ gène, échelles = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
ERREUR
Error in ggplot(data = moyenne_exp_by_time_sex, mapping = aes(x = temps, : could not find function "ggplot"R
Mean_exp_by_chromosome <- rna %>%
group_by(chromosome_name, time) %>%
summary(mean_exp = Mean(expression_log))
ERREUR
Error in rna %>% group_by(chromosome_name, time) %>% summary(mean_exp = Mean(expression_log)): could not find function "%>%"R
ggplot(data = Mean_exp_by_chromosome, mapping = aes( x = temps,
y = moyenne_exp)) +
geom_line() +
facet_wrap(~ chromosome_name, scales = "free_y")
ERREUR
Error in ggplot(data = Mean_exp_by_chromosome, mapping = aes(x = temps, : could not find function "ggplot"La géométrie facet_wrap extrait les tracés dans un
nombre arbitraire de dimensions pour leur permettre de s’adapter
proprement à une seule page. On the other hand, the
facet_grid geometry allows you to explicitly specify how
you want your plots to be arranged via formula notation
(rows ~ columns; a . can be used as a
placeholder that indicates only one row or column).
Modifions le graphique précédent pour comparer l’évolution de l’expression génétique moyenne des hommes et des femmes au fil du temps :
R
# Une colonne, facette par lignes
ggplot(data = Mean_exp_by_time_sex,
mapping = aes(x = time, y = Mean_exp, color = gene)) +
geom_line() +
facet_grid(sexe ~ .)
ERREUR
Error in ggplot(data = Mean_exp_by_time_sex, mapping = aes(x = time, y = Mean_exp, : could not find function "ggplot"R
# Une ligne, facette par colonne
ggplot(data = Mean_exp_by_time_sex,
mapping = aes(x = time, y = Mean_exp, color = gene)) +
geom_line() +
facet_grid(. ~ sexe)
ERREUR
Error in ggplot(data = Mean_exp_by_time_sex, mapping = aes(x = time, y = Mean_exp, : could not find function "ggplot"Thèmes ggplot2
En plus de theme_bw(), qui change l’arrière-plan de
l’intrigue en blanc, ggplot2 est livré avec plusieurs
autres thèmes qui peuvent être utiles pour changer rapidement
l’apparence de votre visualisation. La liste complète des thèmes est
disponible sur https://ggplot2.tidyverse.org/reference/ggtheme.html.
theme_minimal() et theme_light() sont
populaires, et theme_void() peut être utile comme point de
départ pour créer un nouveau thème créé à la main.
Le package ggthemes
fournit une grande variété d’options (y compris un thème Excel 2003 ).
The ggplot2 provides a list of packages that extend the capabilities of
ggplot2, including additional themes.
Personnalisation
Revenons à l’intrigue à facettes de l’expression moyenne par temps et gène, colorée par sexe.
Jetez un œil à la feuille de triche , et réfléchissez aux moyens vous pourriez améliorer l’intrigue.
Maintenant, nous pouvons changer les noms des axes en quelque chose de plus informatif que ‘time’ et ‘mean_exp’, et ajouter un titre à la figure :
R
ggplot(data = moyenne_exp_by_time_sex,
mapping = aes(x = temps, y = moyenne_exp, couleur = sexe)) +
geom_line() +
facet_wrap(~ gène, échelles = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "Expression moyenne des gènes selon la durée de l'infection",
x = "Durée de l'infection (en jours)",
y = "Expression moyenne des gènes")
ERREUR
Error in ggplot(data = moyenne_exp_by_time_sex, mapping = aes(x = temps, : could not find function "ggplot"Les axes ont des noms plus informatifs, mais leur lisibilité peut être améliorée en augmentant la taille de la police :
R
ggplot(data = moyenne_exp_by_time_sex,
mapping = aes(x = temps, y = moyenne_exp, couleur = sexe)) +
geom_line() +
facet_wrap(~ gène, échelles = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "Expression moyenne des gènes selon la durée de l'infection",
x = "Durée de l'infection (en jours)",
y = "Expression génétique moyenne") +
theme(text = element_text(size = 16))
ERREUR
Error in ggplot(data = moyenne_exp_by_time_sex, mapping = aes(x = temps, : could not find function "ggplot"Notez qu’il est également possible de changer les polices de vos
tracés. Si vous êtes sous Windows, vous devrez peut-être installer le extrafont
package.
Nous pouvons personnaliser davantage la couleur du texte des axes x
et y, la couleur de la grille, etc. Nous pouvons aussi par exemple
déplacer la légende vers le haut en définissant
legend.position sur "top".
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "Mean gene expression by duration of the infection",
x = "Duration of the infection (in days)",
y = "Mean gene expression") +
theme(text = element_text(size = 16),
axis.text.x = element_text(colour = "royalblue4", size = 12),
axis.text.y = element_text(colour = "royalblue4", size = 12),
panel.grid = element_line(colour="lightsteelblue1"),
legend.position = "top")
ERREUR
Error in ggplot(data = mean_exp_by_time_sex, mapping = aes(x = time, y = mean_exp, : could not find function "ggplot"Si vous préférez les modifications que vous avez créées au thème par défaut, vous pouvez les enregistrer en tant qu’objet pour pouvoir les appliquer facilement à d’autres tracés que vous pourriez créer. Voici un exemple avec l’histogramme que nous avons créé précédemment.
R
blue_theme <- theme(axis.text.x = element_text(colour = "royalblue4",
size = 12),
axis.text.y = element_text(colour = "royalblue4",
size = 12),
text = element_text(size = 16),
panel.grid = element_line(colour="lightsteelblue1"))
ERREUR
Error in theme(axis.text.x = element_text(colour = "royalblue4", size = 12), : could not find function "theme"R
ggplot(rna, aes(x = expression_log)) +
geom_histogram(bins = 20) +
blue_theme
ERREUR
Error in ggplot(rna, aes(x = expression_log)): could not find function "ggplot"Défi
Avec toutes ces informations en main, veuillez prendre encore cinq minutes pour soit améliorer l’un des tracés générés dans cet exercice, soit créer votre propre graphique. Utilisez la feuille de triche RStudio ggplot2 pour vous inspirer. Voici quelques idées :
- Voyez si vous pouvez modifier l’épaisseur des lignes.
- Pouvez-vous trouver un moyen de changer le nom de la légende ? Qu’en
est-il de ses étiquettes ? (indice : recherchez une fonction ggplot
commençant par
scale_) - Essayez d’utiliser une palette de couleurs différente ou de spécifier manuellement les couleurs pour les lignes (voir http://www.cookbook-r.com/Graphs/Colors_(ggplot2)/).
Par exemple, sur la base de ce tracé :
R
ggplot(data = moyenne_exp_by_time_sex,
mapping = aes(x = temps, y = moyenne_exp, couleur = sexe)) +
geom_line() +
facet_wrap(~ gène, échelles = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
ERREUR
Error in ggplot(data = moyenne_exp_by_time_sex, mapping = aes(x = temps, : could not find function "ggplot"Nous pouvons le personnaliser des manières suivantes :
R
# change the thickness of the lines
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line(size=1.5) +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
ERREUR
Error in ggplot(data = mean_exp_by_time_sex, mapping = aes(x = time, y = mean_exp, : could not find function "ggplot"R
# change the name of the legend and the labels
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_discrete(name = "Gender", labels = c("F", "M"))
ERREUR
Error in ggplot(data = mean_exp_by_time_sex, mapping = aes(x = time, y = mean_exp, : could not find function "ggplot"R
# using a different color palette
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_brewer(name = "Gender", labels = c("F", "M"), palette = "Dark2")
ERREUR
Error in ggplot(data = mean_exp_by_time_sex, mapping = aes(x = time, y = mean_exp, : could not find function "ggplot"R
# manually specifying the colors
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_manual(name = "Gender", labels = c("F", "M"),
values = c("royalblue", "deeppink"))
ERREUR
Error in ggplot(data = mean_exp_by_time_sex, mapping = aes(x = time, y = mean_exp, : could not find function "ggplot"Composer des intrigues
Faceting is a great tool for splitting one plot into multiple subplots, but sometimes you may want to produce a single figure that contains multiple independent plots, i.e. plots that are based on different variables or even different data frames.
Commençons par créer les deux tracés que nous souhaitons disposer l’un à côté de l’autre :
Le premier graphique compte le nombre de gènes uniques par
chromosome. Nous devons d’abord réorganiser les niveaux de
chromosome_name et filtrer les gènes uniques par
chromosome. Nous modifions également l’échelle de l’axe y en une échelle
log10 pour une meilleure lisibilité.
R
arn$chromosome_name <- factor(rna$chromosome_name,
niveaux = c(1:19,"X","Y"))
ERREUR
Error in factor(rna$chromosome_name, niveaux = c(1:19, "X", "Y")): unused argument (niveaux = c(1:19, "X", "Y"))R
count_gene_chromosome <- rna %> % select(chromosome_name, gene) %>%
distinct() %>% ggplot() +
geom_bar(aes(x = chromosome_name), fill = "seagreen",
position = "esquive", stat = "count") +
labs(y = "log10(n gènes)", x = "chromosome") +
scale_y_log10()
ERREUR
Error in rna %> % select(chromosome_name, gene) %>% distinct() %>% ggplot(): could not find function "%>%"R
count_gene_chromosome
ERREUR
Error in eval(expr, envir, enclos): object 'count_gene_chromosome' not foundCi-dessous, nous supprimons également complètement la légende en
définissant legend.position sur "none".
R
exp_boxplot_sex <- ggplot(rna, aes(y=expression_log, x = as.factor(time),
color=sex)) +
geom_boxplot(alpha = 0) +
labs(y = "Exp moyenne du gène",
x = "time") + theme(legend.position = "none")
ERREUR
Error in ggplot(rna, aes(y = expression_log, x = as.factor(time), color = sex)): could not find function "ggplot"R
exp_boxplot_sex
ERREUR
Error in eval(expr, envir, enclos): object 'exp_boxplot_sex' not foundLe package patchwork
fournit une approche élégante pour combiner des figures en utilisant le
« + » pour disposer les figures (généralement latérales). de côté). Plus
précisément, le | les dispose explicitement côte à côte et
/ les empile les uns sur les autres .
R
install.packages("patchwork")
R
library("patchwork")
ERREUR
Error in library("patchwork"): there is no package called 'patchwork'R
count_gene_chromosome + exp_boxplot_sex
ERREUR
Error in eval(expr, envir, enclos): object 'count_gene_chromosome' not foundR
## ou count_gene_chromosome | exp_boxplot_sex
R
count_gene_chromosome / exp_boxplot_sex
ERREUR
Error in eval(expr, envir, enclos): object 'count_gene_chromosome' not foundNous pouvons combiner un contrôle plus approfondi de la mise en page
de la composition finale avec plot_layout pour créer des
mises en page plus complexes :
R
count_gene_chromosome + exp_boxplot_sex + plot_layout(ncol = 1)
ERREUR
Error in eval(expr, envir, enclos): object 'count_gene_chromosome' not foundR
count_gene_chromosome +
(count_gene_chromosome + exp_boxplot_sex) +
exp_boxplot_sex +
plot_layout(ncol = 1)
ERREUR
Error in eval(expr, envir, enclos): object 'count_gene_chromosome' not foundLe dernier tracé peut également être créé à l’aide des compositeurs
| et / :
R
count_gene_chromosome /
(count_gene_chromosome | exp_boxplot_sex) /
exp_boxplot_sex
ERREUR
Error in eval(expr, envir, enclos): object 'count_gene_chromosome' not foundApprenez-en plus sur patchwork sur sa page Web ou dans cette
vidéo.
Une autre option est le package
gridExtra qui permet de combiner des
ggplots séparés en une seule figure en utilisant
grid.arrange() :
R
install.packages("gridExtra")
R
library("gridExtra")
ERREUR
Error in library("gridExtra"): there is no package called 'gridExtra'R
grid.arrange(count_gene_chromosome, exp_boxplot_sex, ncol = 2)
ERREUR
Error in grid.arrange(count_gene_chromosome, exp_boxplot_sex, ncol = 2): could not find function "grid.arrange"En plus des arguments ncol et nrow,
utilisés pour créer des arrangements simples, il existe des outils pour
construire
des dispositions plus complexes.
Exporter des tracés
Après avoir créé votre tracé, vous pouvez l’enregistrer dans un fichier dans votre format préféré. L’onglet Exporter dans le volet Plot de RStudio enregistrera vos tracés à basse résolution, ce qui ne sera pas accepté par de nombreuses revues et ne s’adaptera pas bien aux affiches.
Utilisez plutôt la fonction ggsave(), qui vous permet de
modifier facilement la dimension et la résolution de votre tracé en
ajustant les arguments appropriés (width,
height et dpi ).
Assurez-vous d’avoir le dossier fig_output/ dans votre
répertoire de travail.
R
my_plot <- ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
labs(title = "Mean gene expression by duration of the infection",
x = "Duration of the infection (in days)",
y = "Mean gene expression") +
guides(color=guide_legend(title="Gender")) +
theme_bw() +
theme(axis.text.x = element_text(colour = "royalblue4", size = 12),
axis.text.y = element_text(colour = "royalblue4", size = 12),
text = element_text(size = 16),
panel.grid = element_line(colour="lightsteelblue1"),
legend.position = "top")
ggsave("fig_output/mean_exp_by_time_sex.png", my_plot, width = 15,
height = 10)
# This also works for grid.arrange() plots
combo_plot <- grid.arrange(count_gene_chromosome, exp_boxplot_sex,
ncol = 2, widths = c(4, 6))
ggsave("fig_output/combo_plot_chromosome_sex.png", combo_plot,
width = 10, dpi = 300)
Remarque : Les paramètres « largeur » et « hauteur » déterminent également la taille de la police dans le tracé enregistré.
Autres packages pour la visualisation
ggplot2 est un package très puissant qui s’intègre très
bien dans notre pipeline tidy data et tidy tools. Il
existe d’autres packages de visualisation dans R qui ne doivent pas être
ignorés.
Graphiques de base
Le système graphique par défaut fourni avec R, souvent appelé
base R graphiques est simple et rapide. Il est basé sur le
*modèle de peintre ou de toile *, où différentes sorties sont
directement superposées les unes sur les autres (voir figure @ref(fig:paintermodel)). This is a fundamental
difference with ggplot2 (and with lattice,
described below), that returns dedicated objects, that are rendered on
screen or in a file, and that can even be updated.
R
par(mfrow = c(1, 3))
plot(1:20, main = "Première couche, produite avec plot(1:20)")
plot(1:20, main = "Une ligne rouge horizontale, ajoutée avec abline(h = 10)")
abline(h = 10, col = "red")
plot(1:20, main = "Un rectangle , ajouté avec rect(5, 5, 15, 15)")
abline(h = 10, col = "red")
rect(5, 5, 15, 15, lwd = 3 )
Une autre différence principale est que la fonction de traçage des
graphiques de base essaie de faire la bonne chose en fonction
de leur type d’entrée, c’est-à-dire qu’ils adapteront leur comportement
en fonction de la classe de leur entrée. C’est encore une fois très
différent de ce que nous avons dans ggplot2, qui n’accepte
que les trames de données en entrée, et qui nécessite que les tracés
soient construits petit à petit.
R
par(mfrow = c(2, 2))
boxplot(rnorm(100),
main = "Boxplot de rnorm(100)")
boxplot(matrix(rnorm( 100), ncol = 10),
main = "Boxplot de la matrice(rnorm(100), ncol = 10)")
hist(rnorm(100))
hist( matrice(rnorm(100), ncol = 10))
ERREUR
Error in matrice(rnorm(100), ncol = 10): could not find function "matrice"
L’approche prête à l’emploi dans les graphiques de base peut être
très efficace pour des figures simples et standards, qui peuvent être
produites très rapidement avec une une seule ligne de code et une seule
fonction telle que plot, ou hist, ou
boxplot, … Les valeurs par défaut ne sont cependant pas
toujours les plus attractives et le réglage des figures, surtout
lorsqu’elles deviennent plus complexes (par exemple pour produire des
facettes), peut devenir long et fastidieux.
Le paquet treillis
Le package lattice est similaire à
ggplot2 dans le sens où il utilise des trames de données en
entrée, renvoie des objets graphiques et prend en charge le facettage.
treillis cependant n’est pas basé sur la grammaire des
graphiques et a une interface plus alambiquée.
Une bonne référence pour le package lattice est @latticebook.
Content from Prochaines étapes
Dernière mise à jour le 2024-09-12 | Modifier cette page
Durée estimée : 90 minutes
Vue d'ensemble
Questions
- Qu’est-ce qu’une « expérience résumée » ?
- Qu’est-ce qu’un bioconducteur ?
Objectifs
- Présentez le projet Bioconducteur.
- Introduire la notion de conteneurs de données.
- Donnez un aperçu du
SummarizedExperiment, largement utilisé dans les analyses omiques.
Prochaines étapes
ERREUR
Error in bibliothèque("tidyverse"): could not find function "bibliothèque"Les données en bioinformatique sont souvent complexes. Pour résoudre ce problème, les développeurs définissent des conteneurs de données spécialisés (appelés classes) qui correspondent aux propriétés des données qu’ils doivent gérer.
Cet aspect est au cœur du projet Bioconductor1 qui utilise la même infrastructure de données de base dans tous les packages. Ce a certainement contribué au succès de Bioconductor. Il est conseillé aux développeurs du package Bioconductor d’utiliser l’infrastructure existante pour assurer la cohérence, l’interopérabilité et la stabilité du projet dans son ensemble .
Pour illustrer un tel conteneur de données omiques, nous présenterons
la classe SummarizedExperiment.
Expérience résumée
La figure ci-dessous représente l’anatomie de la classe SummarizedExperiment.

Les objets de la classe SummarizedExperiment contiennent :
Un (ou plusieurs) test(s) contenant les données omiques quantitatives (données d’expression), stockées sous forme d’objet de type matriciel. Caractéristiques (gènes, transcrits, protéines, …) sont définis le long des lignes, et les échantillons le long des colonnes.
Un emplacement exemple de métadonnées contenant des exemples de covariables, stocké sous forme de trame de données . Les lignes de ce tableau représentent des échantillons (les lignes correspondent exactement aux colonnes des données d’expression).
Un emplacement de métadonnées de fonctionnalité contenant des covariables de fonctionnalité, stockées sous forme de une trame de données. Les lignes de ce bloc de données correspondent exactement aux lignes des données d’expression .
La nature coordonnée du SummarizedExperiment garantit
que lors de la manipulation des données, les dimensions des différents
emplacements seront toujours (c’est-à-dire les colonnes des données
d’expression puis les lignes de les exemples de métadonnées, ainsi que
les lignes des données d’expression et les métadonnées des
fonctionnalités) lors de la manipulation des données. Par exemple, si
nous devions exclure un échantillon du test, il serait automatiquement
supprimé des métadonnées de l’échantillon au cours de la même
opération.
Les emplacements de métadonnées peuvent développer des co-variables supplémentaires (colonnes) sans affecter les autres structures.
Création d’une expérience résumée
Afin de créer un SummarizedExperiment, nous allons créer
les composants individuels, c’est-à-dire la matrice de comptage,
l’échantillon et le gène métadonnées à partir de fichiers csv. C’est
généralement ainsi que les données RNA-Seq sont fournies (après le
traitement des données brutes).
ERREUR
Error in read_csv("data/rnaseq.csv"): could not find function "read_csv"ERREUR
Error in rna %>% select(gene, sample, expression) %>% pivot_wider(names_from = sample, : could not find function "%>%"ERREUR
Error in counts %>% select(-gene) %>% as.matrix(): could not find function "%>%"ERREUR
Error in eval(expr, envir, enclos): object 'counts' not foundERREUR
Error in rna %>% select(sample, organism, age, sex, infection, strain, : could not find function "%>%"ERREUR
Error in eval(expr, envir, enclos): object 'sample_metadata' not foundERREUR
Error in rna %>% select(gene, ENTREZID, product, ensembl_gene_id, external_synonym, : could not find function "%>%"ERREUR
Error in eval(expr, envir, enclos): object 'gene_metadata' not foundERREUR
Error in eval(expr, p): object 'count_matrix' not foundERREUR
Error in eval(expr, p): object 'gene_metadata' not foundERREUR
Error in eval(expr, p): object 'sample_metadata' not found-
Une matrice d’expression : nous chargeons la
matrice de comptage, en spécifiant que les premières colonnes
contiennent des noms de lignes/gènes, et convertissons le
data.frameen unematrice. Vous pouvez le télécharger ici.
R
count_matrix <- read.csv("data/count_matrix.csv",
row.names = 1) %>%
as.matrix()
ERREUR
Error in read.csv("data/count_matrix.csv", row.names = 1) %>% as.matrix(): could not find function "%>%"R
count_matrix[1:5, ]
ERREUR
Error in eval(expr, envir, enclos): object 'count_matrix' not foundR
dim(count_matrix)
ERREUR
Error in eval(expr, envir, enclos): object 'count_matrix' not found- Un tableau décrivant les échantillons, disponible ici.
R
sample_metadata <- read.csv("data/sample_metadata.csv")
sample_metadata
SORTIE
sample organism age sex infection strain time tissue mouse
1 GSM2545336 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 14
2 GSM2545337 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 9
3 GSM2545338 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 10
4 GSM2545339 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 15
5 GSM2545340 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 18
6 GSM2545341 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum 6
7 GSM2545342 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 5
8 GSM2545343 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum 11
9 GSM2545344 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 22
10 GSM2545345 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 13
11 GSM2545346 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum 23
12 GSM2545347 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum 24
13 GSM2545348 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 8
14 GSM2545349 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum 7
15 GSM2545350 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 1
16 GSM2545351 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 16
17 GSM2545352 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 21
18 GSM2545353 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 4
19 GSM2545354 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum 2
20 GSM2545362 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 20
21 GSM2545363 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 12
22 GSM2545380 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 19R
dim(sample_metadata)
SORTIE
[1] 22 9- Un tableau décrivant les gènes, disponible ici.
R
gene_metadata <- read.csv("data/gene_metadata.csv")
gene_metadata[1:10, 1:4]
SORTIE
gene ENTREZID
1 Asl 109900
2 Apod 11815
3 Cyp2d22 56448
4 Klk6 19144
5 Fcrls 80891
6 Slc2a4 20528
7 Exd2 97827
8 Gjc2 118454
9 Plp1 18823
10 Gnb4 14696
product
1 argininosuccinate lyase, transcript variant X1
2 apolipoprotein D, transcript variant 3
3 cytochrome P450, family 2, subfamily d, polypeptide 22, transcript variant 2
4 kallikrein related-peptidase 6, transcript variant 2
5 Fc receptor-like S, scavenger receptor, transcript variant X1
6 solute carrier family 2 (facilitated glucose transporter), member 4
7 exonuclease 3'-5' domain containing 2
8 gap junction protein, gamma 2, transcript variant 1
9 proteolipid protein (myelin) 1, transcript variant 1
10 guanine nucleotide binding protein (G protein), beta 4, transcript variant X2
ensembl_gene_id
1 ENSMUSG00000025533
2 ENSMUSG00000022548
3 ENSMUSG00000061740
4 ENSMUSG00000050063
5 ENSMUSG00000015852
6 ENSMUSG00000018566
7 ENSMUSG00000032705
8 ENSMUSG00000043448
9 ENSMUSG00000031425
10 ENSMUSG00000027669R
dim(gene_metadata)
SORTIE
[1] 1474 9Nous allons créer un SummarizedExperiment à partir de
ces tables :
La matrice de comptage qui sera utilisée comme
essaiLe tableau décrivant les échantillons sera utilisé comme emplacement de métadonnées **sample **
Le tableau décrivant les gènes sera utilisé comme emplacement de métadonnées **features **
Pour ce faire, nous pouvons assembler les différentes parties à
l’aide du constructeur SummarizedExperiment :
R
## BiocManager::install("SummarizedExperiment")
bibliothèque("SummarizedExperiment")
ERREUR
Error in bibliothèque("SummarizedExperiment"): could not find function "bibliothèque"Tout d’abord, nous nous assurons que les échantillons sont dans le même ordre dans la matrice de comptage et l’annotation d’échantillon, et il en va de même pour les gènes dans la matrice de comptage et l’annotation des gènes.
R
stopifnot(rownames(count_matrix) == gene_metadata$gene)
ERREUR
Error in eval(expr, envir, enclos): object 'count_matrix' not foundR
stopifnot(colnames(count_matrix) == sample_metadata$sample)
ERREUR
Error in eval(expr, envir, enclos): object 'count_matrix' not foundR
se <- SummarizedExperiment(assays = list(counts = count_matrix),
colData = sample_metadata,
rowData = gene_metadata)
ERREUR
Error in SummarizedExperiment(assays = list(counts = count_matrix), colData = sample_metadata, : could not find function "SummarizedExperiment"R
se
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundLa sauvegarde des données
L’export de données vers un tableur, comme nous l’avons fait dans un
épisode précédent, présente plusieurs limitations, comme celles décrites
dans le premier chapitre (éventuelles incohérences avec ,
et . pour les séparateurs décimaux et manque de définitions
de types de variables). De plus, l’exportation de données vers une
feuille de calcul n’est pertinente que pour les données rectangulaires
telles que les dataframes et les matrices.
Une manière plus générale de sauvegarder des données, spécifique à R
et dont le fonctionnement est garanti sur n’importe quel système
d’exploitation, consiste à utiliser la fonction saveRDS .
L’enregistrement d’objets comme celui-ci générera une représentation
binaire sur le disque (en utilisant l’extension de fichier
rds ici), qui peut être rechargée dans R à l’aide de la
fonction readRDS.
R
saveRDS(se, file = "data_output/se.rds")
rm(se)
se <- readRDS("data_output/se.rds")
head(se)
Pour conclure, lorsqu’il s’agit de sauvegarder des données de R qui
seront chargées à nouveau dans R, la sauvegarde et le chargement avec
saveRDS et readRDS sont l’approche préférée.
Si les données tabulaires doivent être partagées avec quelqu’un qui
n’utilise pas R, alors l’exportation vers une feuille de calcul
textuelle est une bonne alternative.
En utilisant cette structure de données, nous pouvons accéder à la
matrice d’expression avec la fonction assay :
R
head(essai(se))
ERREUR
Error in essai(se): could not find function "essai"R
dim(essai(se))
ERREUR
Error in essai(se): could not find function "essai"Nous pouvons accéder aux exemples de métadonnées à l’aide de la
fonction colData :
R
colData(se)
ERREUR
Error in colData(se): could not find function "colData"R
dim(colData(se))
ERREUR
Error in colData(se): could not find function "colData"Nous pouvons également accéder aux métadonnées des fonctionnalités à
l’aide de la fonction rowData :
R
head(rowData(se))
ERREUR
Error in rowData(se): could not find function "rowData"R
dim(rowData(se))
ERREUR
Error in rowData(se): could not find function "rowData"Sous-ensemble d’une expérience résumée
SummarizedExperiment peut être un sous-ensemble comme avec des trames de données, avec des chiffres ou avec des caractères logiques.
Ci-dessous, nous créons une nouvelle instance de la classe SummarizedExperiment qui contient uniquement les 5 premières fonctionnalités pour les 3 premiers échantillons.
R
se1 <- se[1:5, 1:3]
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundR
se1
ERREUR
Error in eval(expr, envir, enclos): object 'se1' not foundR
colData(se1)
ERREUR
Error in colData(se1): could not find function "colData"R
rowData(se1)
ERREUR
Error in rowData(se1): could not find function "rowData"Nous pouvons également utiliser la fonction colData()
pour créer un sous-ensemble sur quelque chose de les exemples de
métadonnées ou la fonction rowData() pour créer un
sous-ensemble sur quelque chose à partir des métadonnées de
fonctionnalité . Par exemple, nous ne conservons ici que les miARN et
les échantillons non infectés :
R
se1 <- se[rowData(se)$gene_biotype == "miRNA",
colData(se)$infection == "NonInfected"]
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundR
se1
ERREUR
Error in eval(expr, envir, enclos): object 'se1' not foundR
assay(se1)
ERREUR
Error in assay(se1): could not find function "assay"R
colData(se1)
ERREUR
Error in colData(se1): could not find function "colData"R
rowData(se1)
ERREUR
Error in rowData(se1): could not find function "rowData"R
assay(se)[1:3, colData(se)$time != 4]
ERREUR
Error in assay(se): could not find function "assay"R
# Equivalent to
assay(se)[1:3, colData(se)$time == 0 | colData(se)$time == 8]
ERREUR
Error in assay(se): could not find function "assay"R
arn |>
filtre(gène %in% c("Asl", "Apod", "Cyd2d22")) |>
filtre(temps != 4) |> select(expression )
ERREUR
Error in select(filtre(filtre(arn, gène %in% c("Asl", "Apod", "Cyd2d22")), : could not find function "select"Le long tableau et le SummarizedExperiment contiennent
les mêmes informations , mais sont simplement structurés différemment.
Chaque approche a ses propres avantages : la première convient bien aux
packages tidyverse, tandis que la seconde est la structure
préférée pour de nombreuses étapes de bioinformatique et de traitement
statistique. Par exemple, une analyse typique d’ARN-Seq utilisant le
package DESeq2.
Ajouter des variables aux métadonnées
Nous pouvons également ajouter des informations aux métadonnées. Supposons que vous souhaitiez ajouter le centre où les échantillons ont été collectés…
R
colData(se)$center <- rep("Université de l'Illinois", nrow(colData(se)))
ERREUR
Error in colData(se): could not find function "colData"R
colData(se)
ERREUR
Error in colData(se): could not find function "colData"Cela illustre que les emplacements de métadonnées peuvent croître indéfiniment sans affecter les autres structures !
TidyRésuméExpérience
Vous vous demandez peut-être si pouvons-nous utiliser les commandes
Tidyverse pour interagir avec les objets
SummarizedExperiment ? La réponse est oui, nous pouvons le
faire avec le package tidySummarizedExperiment.
Rappelez-vous à quoi ressemble notre objet SummarizedExperiment :
R
se
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundChargez tidySummarizedExperiment puis jetez à nouveau un
œil à l’objet se .
R
#BiocManager::install("tidySummarizedExperiment")
library("tidySummarizedExperiment")
ERREUR
Error in library("tidySummarizedExperiment"): there is no package called 'tidySummarizedExperiment'R
se
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundIl s’agit toujours d’un objet SummarizedExperiment, il
conserve donc la structure efficace , mais nous pouvons maintenant le
voir comme un tibble. Notez la première ligne de la sortie dit ceci,
c’est une abstraction
SummarizedExperiment-tibble . Nous pouvons
également voir dans la deuxième ligne de la sortie le nombre de
transcriptions et d’échantillons.
Si nous voulons revenir à la vue standard
SummarizedExperiment, nous pouvons le faire.
R
options("restore_SummarizedExperiment_show" = TRUE)
se
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundMais ici, nous utilisons la vue tibble.
R
options("restore_SummarizedExperiment_show" = FALSE)
se
ERREUR
Error in eval(expr, envir, enclos): object 'se' not foundNous pouvons maintenant utiliser les commandes Tidyverse pour
interagir avec l’objet SummarizedExperiment.
Nous pouvons utiliser filter pour filtrer les lignes en
utilisant une condition, par exemple pour afficher toutes les lignes
pour un échantillon.
R
se %>% filtre(.sample == "GSM2545336")
ERREUR
Error in se %>% filtre(.sample == "GSM2545336"): could not find function "%>%"Nous pouvons utiliser « select » pour spécifier les colonnes que nous voulons afficher.
R
se %>% sélectionner (.sample)
ERREUR
Error in se %>% sélectionner(.sample): could not find function "%>%"Nous pouvons utiliser mutate pour ajouter des
informations sur les métadonnées.
R
se %>% muter(center = "Université de Heidelberg")
ERREUR
Error in se %>% muter(center = "Université de Heidelberg"): could not find function "%>%"Nous pouvons également combiner des commandes avec le tube Tidyverse
%>%. Pour l’exemple de , nous pourrions combiner
group_by et summarise pour obtenir le nombre
total de pour chaque échantillon.
R
se %>%
group_by(.sample) %>%
summarise(total_counts=sum(counts))
ERREUR
Error in se %>% group_by(.sample) %>% summarise(total_counts = sum(counts)): could not find function "%>%"Nous pouvons traiter l’objet SummarizedExperiment bien rangé comme un tibble normal pour le traçage.
Ici, nous traçons la distribution des comptes par échantillon.
R
se %>%
ggplot(aes(counts + 1, group=.sample, color=infection)) +
geom_density() +
scale_x_log10() +
theme_bw()
ERREUR
Error in se %>% ggplot(aes(counts + 1, group = .sample, color = infection)): could not find function "%>%"Pour plus d’informations sur TidySummarizedExperiment, consultez le site Web du packageici.
Message à retenir
SummarizedExperimentreprésente un moyen efficace de stocker et de gérer les données omiques.Ils sont utilisés dans de nombreux packages Bioconductor.
Si vous suivez la prochaine formation axée sur l’analyse de
séquençage d’ARN, vous apprendrez à utiliser le package Bioconductor
DESeq2 pour faire des analyses d’expression différentielle.
L’ensemble de l’analyse du package DESeq2 est géré dans un
SummarizedExperiment.
Le Bioconductor a été initié par Robert Gentleman, l’un des deux créateurs du langage R . Bioconductor fournit des outils dédiés à l’analyse des données omiques . Bioconductor utilise le langage de programmation statistique R et est open source et développement ouvert.↩︎