Differential expression 解析
Last updated on 2026-04-28 | Edit this page
Overview
Questions
- 典型的な Differential expression 解析で実施される主な手順は何ですか?
- DESeq2の出力結果をどのように解釈すればよいですか?
Objectives
- Differential expression 解析における主要な手順について説明してください。
- DESeq2パッケージを使用してR環境でこれらの手順を実行する方法を説明してください。
Differential expression の推定
RNA-seqデータ解析における主要な目的の一つは、実験群間または条件間(例:処理群と対照群、時間点、組織など)における系統的な変化を定量化し、統計的に推論することです。 これは通常、条件間変動と条件内変動を用いて発現変動パターンを示す遺伝子を同定することで行われ、生物学的複製サンプル(同一条件下での複数サンプル)が必要となります。 発現変動解析を実施するためのソフトウェアパッケージは複数存在します。 比較研究によれば、発現変動遺伝子(DEG)に関してはある程度の一致が見られるものの、ツール間にはばらつきがあり、どのツールも他のすべてのツールを一貫して上回る性能を示すことはないことが報告されています(Soneson and Delorenzi, 2013参照)。 以下では、DESeq2 ソフトウェアパッケージを使用した発現変動解析の実施方法を説明し、実際に解析を行います。edgeRパッケージも同様の手法を実装しており、カウントデータに関する主要な仮定を共有しています。 両パッケージとも一般的に良好な安定性を示し、同等の結果が得られます。
DESeqDataSetオブジェクト
DESeq2を実行するには、カウントデータをDESeqDataSetクラスのオブジェクトとして表現する必要があります。
DESeqDataSetはSummarizedExperimentクラス(定量データのインポートとアノテーションセクション参照)の拡張版であり、カウントアッセイデータ、特徴量(ここでは遺伝子)、およびサンプルメタデータに加えて、
design formula を保持します。 design formula
は、モデリング時に用いる変数を表現するものです。通常は解析対象の変数(群変数)や、考慮したいその他の変数(例:バッチ効果変数)などが含まれます。
design formula および関連する design matrices
に関する詳細な説明は、design
matricesの詳細な探索セクションで行います。DESeqDataSetクラスのオブジェクトは、カウント行列、SummarizedExperimentオブジェクト、トランスクリプト存在量ファイル、またはhtseqカウントファイルから構築可能です。
パッケージの読み込み
R
suppressPackageStartupMessages({
library(SummarizedExperiment)
library(DESeq2)
library(ggplot2)
library(ExploreModelMatrix)
library(cowplot)
library(ComplexHeatmap)
library(apeglm)
})
データの読み込み
前回の品質管理分析で使用した SummarizedExperiment
オブジェクトを再度読み込みます。品質管理の探索的分析では、カウント数が5未満の遺伝子約35%を削除しました。これらの遺伝子は情報量が不足していたためです。DESeq2の統計解析においては、デフォルトで独立したフィルタリングが行われるため、これらの遺伝子を厳密に削除する必要はありません。ただし、これにより
DESeqDataSet
オブジェクトのメモリ使用量が減少し、計算速度が向上する可能性があります。さらに、これらの遺伝子が可視化結果を乱雑にするのを防ぐことができます。
R
se <- readRDS("data/GSE96870_se.rds")
se <- se[rowSums(assay(se, "counts")) > 5, ]
DESeqDataSetの作成
本例で使用する design matrix は ~ sex + time
とします。
This will allow us test the difference between males and females (averaged over time point) and the difference between day 0, 4 and 8 (averaged over males and females). If we wanted to test other comparisons (e.g., Female.Day8 vs. Female.Day0 and also Male.Day8 vs. Male.Day0) we could use a different design matrix to more easily extract those pairwise comparisons.
R
dds <- DESeq2::DESeqDataSet(se,
design = ~ sex + time)
WARNING
Warning in DESeq2::DESeqDataSet(se, design = ~sex + time): some variables in
design formula are characters, converting to factors正規化処理
DESeq2 と edgeR
は以下の前提条件に基づいています:
- ほとんどの遺伝子は発現量に有意な差がない
- 特定の遺伝子にリードがマッピングされる確率は、同一グループ内のすべてのサンプルにおいて同一である
前のセクションで示したように、サンプルの総カウント数は(たとえ同じ条件であっても)ライブラリサイズ(シーケンスされたリードの総数)に依存します。 特定の遺伝子のカウント数の変動をグループ間およびグループ内で比較するには、まずライブラリサイズと構成効果を考慮する必要があります。 前のセクションで説明したestimateSizeFactors()関数を思い出してください。
R
dds <- estimateSizeFactors(dds)
統計モデリング
DESeq2 と edgeR は、グループあたりのレプリケートが少ないこと、平均分散依存性 (探索的データ分析を参照)、および歪んだカウント分布を考慮して、RNA-seq カウントを 負の二項分布 としてモデル化します。
分散パラメータ (Dispersion)
負の二項分布に従う遺伝子のカウント値におけるグループ内分散は、平均 \(\mu\) に対して以下のようにモデル化できます:
\(var = \mu + \theta \mu^2\)
ここで \(\theta\) は遺伝子固有の
分散パラメータ (dispersion)
を表し、データのばらつき度合いを示す指標です。第二段階として、各遺伝子の分散パラメータを推定することで、グループ内の期待分散値を算出し、グループ間の差異を検定します。サンプル数が限られている場合、適切な分散推定値は得にくいため、類似した発現パターンを示す遺伝子間の情報が活用されます。各遺伝子の分散推定値は、観測された分散分布の中央値方向に「縮小」(shrinked)されます。DESeq2
では、estimateDispersions()
関数を使用して分散推定値を取得できます。 plotDispEsts()
関数を用いることで、この 縮小効果
の影響を視覚的に確認することが可能です:
R
dds <- estimateDispersions(dds)
OUTPUT
gene-wise dispersion estimatesOUTPUT
mean-dispersion relationshipOUTPUT
final dispersion estimatesR
plotDispEsts(dds)
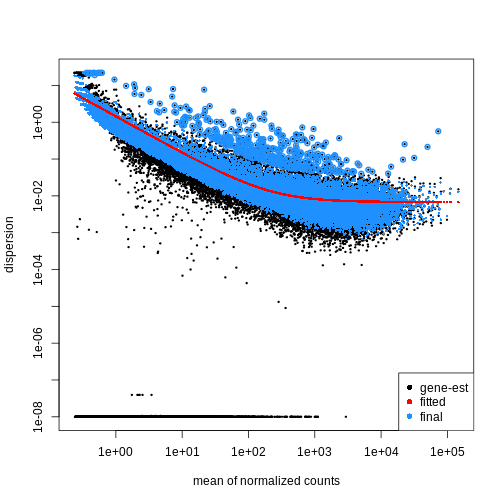
検定方法
We can use the nbinomWaldTest()function of
DESeq2 to fit a generalized linear model (GLM) and
compute log2 fold changes (synonymous with “GLM coefficients”,
“beta coefficients” or “effect size”) corresponding to the variables of
the design matrix. The design matrix is directly
related to the design formula and automatically derived from
it. Assume a design formula with one variable (~ treatment)
and two factor levels (treatment and control). The mean expression \(\mu_{j}\) of a specific gene in sample
\(j\) will be modeled as following:
\(log(μ_j) = β_0 + x_j β_T\),
with \(β_T\) corresponding to the log2 fold change of the treatment groups, \(x_j\) = 1, if \(j\) belongs to the treatment group and \(x_j\) = 0, if \(j\) belongs to the control group.
Finally, the estimated log2 fold changes are scaled by their standard error and tested for being significantly different from 0 using the Wald test.
R
dds <- nbinomWaldTest(dds)
注意
上記で説明した標準的な差異発現解析は、単一の関数 DESeq()
にまとめられています。最初のコードブロックを実行することは、2番目のコードブロックを実行することと等価です:
R
dds <- DESeq(dds)
R
dds <- estimateSizeFactors(dds)
dds <- estimateDispersions(dds)
dds <- nbinomWaldTest(dds)
特定の対比に関する結果の探索
results()
関数を使用すると、遺伝子ごとの検定統計量(対数2倍変化量や調整済みp値など)を抽出できます。比較対象とする対比は、モデル係数の線形結合として定義します(これは設計行列内の列の組み合わせに相当します)。したがって、この定義は設計式と直接関連しています。設計行列の詳細な解説と、異なる対比を指定するための使用方法については、設計行列の探索
のセクションを参照してください。results()
関数では、対比を対象変数(参照変数)と、その比較対象となる水準を用いて
contrast
引数で指定します。デフォルトでは、参照変数は設計式の最後の変数となり、参照水準
は最初の因子水準、最後の水準
が比較対象として自動的に使用されます。また、results()
関数の name
引数を使用して明示的に対比を指定することも可能です。利用可能なすべての対比名は、resultsNames()
関数で取得できます。
Challenge
このレッスンで使用されている例に対して results(dds)
を実行する際、比較のデフォルト値となる
コントラスト、基準レベル、および
「最終レベル」 はそれぞれ何になりますか?
ヒント:オブジェクト構築に使用された設計式を確認してください。
このレッスンの例では、設計式の最後の変数は time です。
基準レベル(アルファベット順で最初の値)は
Day0 で、最終レベルは Day8
です。
R
levels(dds$time)
OUTPUT
[1] "Day0" "Day4" "Day8"デフォルトの対比を特定するのが難しい場合でも、results()
関数の出力には明示的に参照している対比が記載されています(以下参照)!
results()関数の出力を詳細に分析するには、summary()関数を使用し、有意性(p値)に基づいて結果を並べ替えることができます。ここでは、特に時間(「関心のある変数」)における変化に関心があり、具体的にはDay0(「基準レベル」)とDay8(「比較対象レベル」)の間で発現量に差異が見られる遺伝子を対象としています。使用したモデルには性別変数が含まれています(上記参照)。したがって、本分析結果は性別に関連する差異を考慮した「補正済み」の値となります。
R
## Day 8 vs Day 0
resTime <- results(dds, contrast = c("time", "Day8", "Day0"))
summary(resTime)
OUTPUT
out of 27430 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4472, 16%
LFC < 0 (down) : 4282, 16%
outliers [1] : 10, 0.036%
low counts [2] : 3723, 14%
(mean count < 1)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
# View(resTime)
head(resTime[order(resTime$pvalue), ])
OUTPUT
log2 fold change (MLE): time Day8 vs Day0
Wald test p-value: time Day8 vs Day0
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
Asl 701.343 1.117332 0.0594128 18.8062 6.71212e-79
Apod 18765.146 1.446981 0.0805056 17.9737 3.13229e-72
Cyp2d22 2550.480 0.910202 0.0556002 16.3705 3.10712e-60
Klk6 546.503 -1.671897 0.1057395 -15.8115 2.59339e-56
Fcrls 184.235 -1.947016 0.1277235 -15.2440 1.80488e-52
A330076C08Rik 107.250 -1.749957 0.1155125 -15.1495 7.63434e-52
padj
<numeric>
Asl 1.59057e-74
Apod 3.71130e-68
Cyp2d22 2.45431e-56
Klk6 1.53639e-52
Fcrls 8.55406e-49
A330076C08Rik 3.01518e-48Challenge
時間の影響を排除して、雄個体と雌個体間における DE 遺伝子を探索してください。
ヒント: GLM
を再度適合させる必要はありません。resultsNames()
関数を使用して正しい対比を取得してください。
R
## Male vs Female
resSex <- results(dds, contrast = c("sex", "Male", "Female"))
summary(resSex)
OUTPUT
out of 27430 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 51, 0.19%
LFC < 0 (down) : 70, 0.26%
outliers [1] : 10, 0.036%
low counts [2] : 8504, 31%
(mean count < 6)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
head(resSex[order(resSex$pvalue), ])
OUTPUT
log2 fold change (MLE): sex Male vs Female
Wald test p-value: sex Male vs Female
DataFrame with 6 rows and 6 columns
baseMean log2FoldChange lfcSE stat pvalue
<numeric> <numeric> <numeric> <numeric> <numeric>
Xist 22603.0359 -11.60429 0.336282 -34.5076 6.16852e-261
Ddx3y 2072.9436 11.87241 0.397493 29.8683 5.08722e-196
Eif2s3y 1410.8750 12.62513 0.565194 22.3377 1.58997e-110
Kdm5d 692.1672 12.55386 0.593607 21.1484 2.85293e-99
Uty 667.4375 12.01728 0.593573 20.2457 3.87772e-91
LOC105243748 52.9669 9.08325 0.597575 15.2002 3.52699e-52
padj
<numeric>
Xist 1.16684e-256
Ddx3y 4.81149e-192
Eif2s3y 1.00253e-106
Kdm5d 1.34915e-95
Uty 1.46702e-87
LOC105243748 1.11194e-48多重検定補正
遺伝子ごとに1回ずつ行われる多数の検定を実施するため、DE解析の結果には必然的に偽陽性が相当数含まれることになります。例えば、20,000個の遺伝子を\(\alpha = 0.05\)の有意水準で検定した場合、実際には発現差が認められない遺伝子が1,000個有意に検出されると予想されます。
このような高い偽陽性率を考慮するため、我々は結果に対して多重検定補正を適用します。DESeq2ではデフォルトで、Benjamini-Hochberg法を用いて、DE解析結果に対する調整済みp値(padj)を算出します。
独立フィルタリングと対数倍率変化の縮小処理
結果はさまざまな方法で可視化できます。特に有用なのは、log2倍変化量、有意に発現変動した遺伝子、および_遺伝子の平均カウント数_の関係性を分析することです。
DESeq2にはこの分析を行うための便利な関数plotMA()が用意されています。
R
plotMA(resTime)
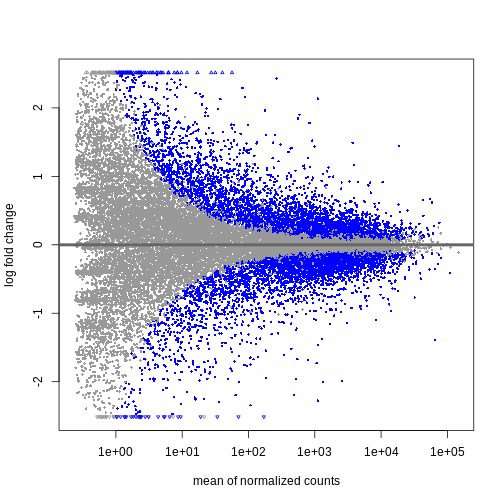
この結果から、平均カウント数が少ない遺伝子ほど対数倍率変化量が大きくなる傾向が確認できます。 これは、発現量の低い遺伝子のカウントデータが非常にノイズを含む性質によるものです。 平均カウント数が少なく分散が大きいこれらの遺伝子については、情報量が限られているため、対数倍率変化量を縮小処理することができます。
R
resTimeLfc <- lfcShrink(dds, coef = "time_Day8_vs_Day0", res = resTime)
OUTPUT
using 'apeglm' for LFC shrinkage. If used in published research, please cite:
Zhu, A., Ibrahim, J.G., Love, M.I. (2018) Heavy-tailed prior distributions for
sequence count data: removing the noise and preserving large differences.
Bioinformatics. https://doi.org/10.1093/bioinformatics/bty895R
plotMA(resTimeLfc)
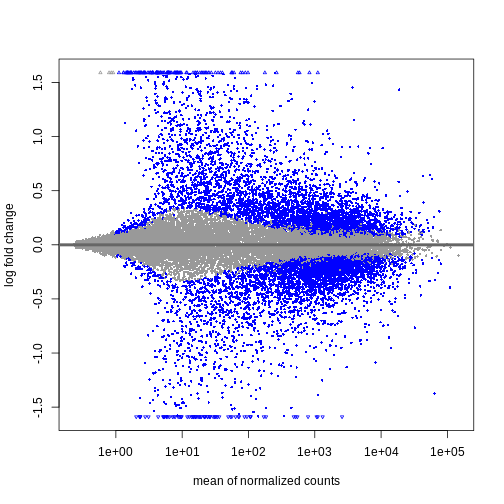
対数倍率変化量の縮小処理は、遺伝子の可視化やランキングにおいて有用ですが、結果の詳細な検討を行う場合には、通常independentFiltering引数を使用して発現量の低い遺伝子を除去します。
Challenge
デフォルトでは independentFiltering は TRUE
に設定されています。低発現遺伝子をフィルタリングしない場合、どのような結果が得られるでしょうか?
summary()
関数を使用して結果を比較してみてください。ほとんどの低発現遺伝子は、有意な発現差を示していません(上記の
MA
プロットでは青色で表示されています)。それでは、結果に差異が生じる原因は何でしょうか?
R
resTimeNotFiltered <- results(dds,
contrast = c("time", "Day8", "Day0"),
independentFiltering = FALSE)
summary(resTime)
OUTPUT
out of 27430 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4472, 16%
LFC < 0 (down) : 4282, 16%
outliers [1] : 10, 0.036%
low counts [2] : 3723, 14%
(mean count < 1)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?resultsR
summary(resTimeNotFiltered)
OUTPUT
out of 27430 with nonzero total read count
adjusted p-value < 0.1
LFC > 0 (up) : 4324, 16%
LFC < 0 (down) : 4129, 15%
outliers [1] : 10, 0.036%
low counts [2] : 0, 0%
(mean count < 0)
[1] see 'cooksCutoff' argument of ?results
[2] see 'independentFiltering' argument of ?results発現頻度が極めて低い遺伝子では、分散が大きいため有意差が認められることはほとんどありません。したがって、低発現遺伝子をフィルタリングすることで、実験全体における偽陽性率を一定に保ったまま検出感度を向上させることが可能となります。
選択した遺伝子セットの可視化
発現変動遺伝子の数は非常に膨大であり、単に発現変動遺伝子をランク付けしたリストだけでは、実験的な疑問に対する解釈が難しい場合があります。遺伝子発現パターンを可視化することで、発現パターンの傾向や、関連する機能を持つ遺伝子群を特定することが可能になります。具体的な遺伝子群の過剰表現については、後のセクションで体系的な検出を行います。ただし、この可視化作業を行うだけでも、今後の解析で期待すべき結果のイメージを掴む上で大いに役立ちます。
可視化には、変換処理を施したデータ(探索的データ解析を参照)と、最も有意に発現変動が認められた遺伝子群を使用します。ヒートマップを作成することで、サンプル群間(列方向)の発現パターンを明らかにできるほか、遺伝子(行方向)をその類似性に基づいて自動的に順序付けすることが可能です。
R
# Transform counts
vsd <- vst(dds, blind = TRUE)
# Get top DE genes
genes <- resTime[order(resTime$pvalue), ] |>
head(10) |>
rownames()
heatmapData <- assay(vsd)[genes, ]
# Scale counts for visualization
heatmapData <- t(scale(t(heatmapData)))
# Add annotation
heatmapColAnnot <- data.frame(colData(vsd)[, c("time", "sex")])
heatmapColAnnot <- HeatmapAnnotation(df = heatmapColAnnot)
# Plot as heatmap
ComplexHeatmap::Heatmap(heatmapData,
top_annotation = heatmapColAnnot,
cluster_rows = TRUE, cluster_columns = FALSE)
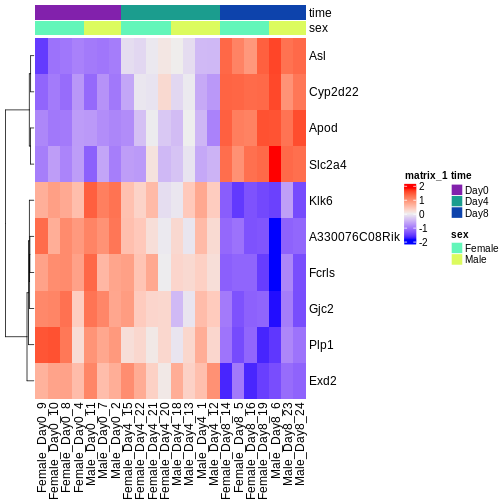
Challenge
ヒートマップと上位のDE遺伝子を確認してください。3時点すべてにおける変化の傾向として、予想通りの結果や予想外の結果は見つかりましたか?
結果の出力
R環境外で独立して使用できる形式の出力ファイルを生成したい場合があります。resTimeオブジェクトの形式は行名として遺伝子シンボルのみを含んでいるため、遺伝子アノテーション情報を結合した上で、.csvファイルとして保存します:
R
head(as.data.frame(resTime))
head(as.data.frame(rowRanges(se)))
temp <- cbind(as.data.frame(rowRanges(se)),
as.data.frame(resTime))
write.csv(temp, file = "output/Day8vsDay0.csv")
- DESeq2では、差異的発現解析の主要な手順(サイズ因子推定、分散推定、検定統計量の算出)が単一の関数DESeq()に統合されています。
- 発現量の低い遺伝子を独立してフィルタリングすることは、多くの場合有効な手法です。