RNA-seq入門
Last updated on 2025-11-11 | Edit this page
Estimated time 100 minutes
Overview
Questions
- RNA-seq実験を計画する際に考慮すべきさまざまな選択とは?
- どのように生のfastqファイルを処理して、遺伝子ごと、サンプルごとのリードカウントの表を作成するのですか?
- ある生物についてアノテーションされた遺伝子の情報はどこで見つけることができますか?
- RNA-seq解析の典型的なステップとは?
Objectives
- RNA-seqとは何か?
- RNA-seq実験を実施する前に行わなければならない最も一般的なデザイン選択のいくつかを説明する。
- 生データから下流の解析に使用されるリードカウントマトリックスまでの手順の概要を説明する。
- RNA-seq解析で生成されるいくつかの一般的なタイプの結果と可視化を示す。
RNA-seq実験では何を測定するのか?
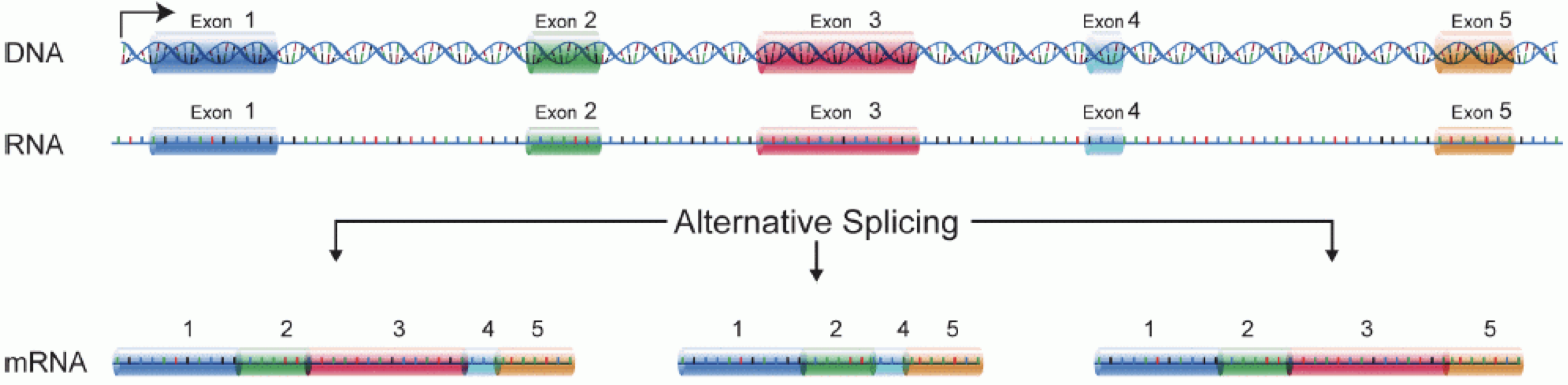
RNA分子を作るには、まずDNAがmRNAに転写される。 その後、イントロン領域がスプライシングされ、エキソン領域が組み合わされて遺伝子の異なる_isoforms_となる。
(図はMartin & Wang (2011)より引用)。
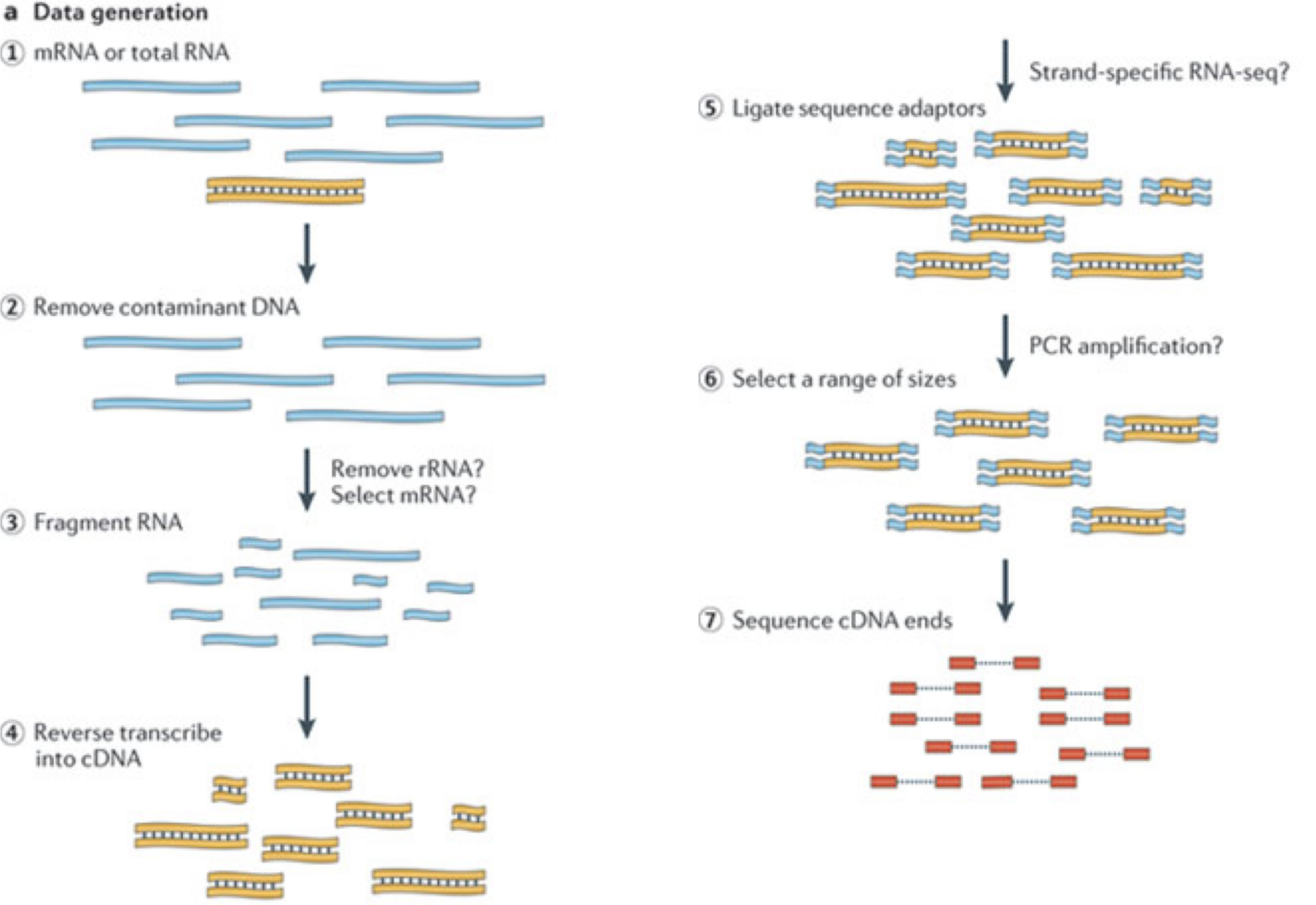
典型的なRNA-seq実験では、まず目的のサンプルからRNA分子を収集する。 ポリA末端を持つ分子(主にmRNA)を濃縮する、あるいは、そうでなければ非常に豊富なリボソームRNAを除去した後、残りの分子を細かく断片化する(分子全体を考慮するロングリード・プロトコルもあるが、このレッスンの焦点ではない)。 イントロン配列を除いたスプライシングのために、RNA分子(したがって生成された断片)はゲノムの途切れることのない領域に対応しない可能性があることを心に留めておくことが重要である。 その後、RNA断片はcDNAに逆転写され、両端に配列決定用アダプターが付加される。 これらのアダプターにより、フラグメントをフローセルに取り付けることができる。 一旦くっつくと、各フラグメントは大きく増幅され、フローセル上に同一配列のクラスターを生成する。 次にシークエンサーは、このようなクラスターに含まれるcDNA断片の最初の50-200塩基の塩基配列を、一端から順に決定する。 多くのデータセットは、両端から断片を読み取る、いわゆるペアエンドプロトコルで作成される。 このようなリード(またはリードのペア)は1回の実験で数百万本生成され、これらは(ペアの)FASTQファイルに表現される。 それぞれのリードは、このようなファイルでは4つの連続した行で表現される。最初に一意のリード識別子の行、次にリードの推定配列、次に別の識別子の行、最後に各推定ヌクレオチドの塩基品質を含む行で、対応する位置のヌクレオチドが正しく同定された確率を表す。
チャレンジ隣人と以下の点について話し合ってください。
- ペアエンドプロトコルの利点と欠点を教えてください。
- リード配列を含むFASTQファイルに対して実行するのに有用な品質評価として、どのようなものが考えられますか?
実験デザインの考察
データ収集を始める前に、実験計画について考える時間を取ることが不可欠である。 実験計画とは、関心のある問題にできるだけ効率的に答えるために、適切な種類のデータと十分な量のデータを確実に入手できるようにすることを目的とした実験の組織に関するものである。 どのような条件や標本群を考慮するか、どれだけの複製を収集するか、実際のデータ収集をどのように計画するかといった側面は、考慮すべき重要な問題である。 多くのハイスループット生物学的実験(RNA-seqを含む)は環境条件に敏感であり、異なる日に、異なる分析者によって、異なるセンターで、異なるバッチの試薬を用いて行われた測定を直接比較することはしばしば困難である。 このため、実験を適切にデザインし、異なるタイプの(一次的効果と二次的効果を)区別できるようにすることが非常に重要である。

(図はLazic (2017)より)。
チャレンジ隣人と話し合う
- なぜ複製が必要なのですか?
重要なのは、統計学的な観点から見ると、すべての反復が同じように有用というわけではないということである。 後者は通常、測定装置の再現性をテストするために使用され、一方、生物学的な複製は、対象集団からの異なるサンプル間のばらつきについて情報を提供する。 別の方式では、複製(または単位)を「生物学的」、「実験的」、「観察的」に分類する。 ここでいう生物学的単位とは、推論を行いたい実体のことである(動物や人など)。 ある治療法の効果について一般的な見解を述べるには、生物学的単位の再現が必要である。1匹のマウスだけを研究して、マウスの集団に対する薬物の効果について結論を出すことはできない。 実験単位とは、ある処置に独立に割り当てることができる最小の実体である(動物、子、ケージ、井戸など)。 実験単位の再現のみが真の再現となる。 最後に、観測単位とは、測定が行われる実体のことである。
興味のある質問に答える能力に対する実験デザインの影響を調べるために、ConfoundingExplorerパッケージで提供されている対話型アプリケーションを使用する。
チャレンジ
ConfoundingExplorerアプリケーションを起動し、インターフェイスに慣れる。
チャレンジ
- 平衡計画(各バッチの2群間の反復分布が等しい)の場合、バッチ効果の強さを増すとどのような効果がありますか? バッチ効果を調整するかどうかは重要か?
- アンバランスなデザイン(1つのグループのほとんど、またはすべての複製が1つのバッチから得られている)が増えている場合、バッチ効果の強さを増すとどのような影響がありますか? バッチ効果を調整するかどうかは重要か?
RNA-seq定量化:リードからカウントマトリックスまで

シーケンサーからのFASTQファイルに含まれるリード配列は、どの遺伝子や転写産物に由来するかという情報を持っていないため、そのままでは一般的に直接役に立たない。 したがって、最初の処理ステップは、各リードの起源の位置を特定し、これを使用して遺伝子(または個々の転写産物などの別の特徴)に由来するリードの数の推定値を取得しようとするものである。 そして、これを遺伝子の存在量、すなわち発現レベルの代用として用いることができる。 RNA定量パイプラインは数多く存在し、最も一般的なアプローチは主に3つのタイプに分類できる:
リードをゲノムにアライメントし、各遺伝子のエクソン内にマップされたリードの数をカウントする。 これは最もシンプルな方法のひとつだ。 トランスクリプトームのアノテーションが不十分な種については、この方法が望ましい。 例:GRCm39 への
STARアライメント +RsubreadfeatureCountsトランスクリプトームへのリードのアラインメント、トランスクリプトーム発現の定量化、トランスクリプトーム発現を遺伝子発現に要約する。 このアプローチは、正確な定量結果(independent benchmarking)を得ることができる。 、特にDNA汚染のない高品質のサンプルについて。 例GENCODE GRCh38 のトランスクリプトームに対する
rsem-calculate-expression --starとtximportを用いた RSEM 定量化。対応するゲノムをおとりとして、トランスクリプトームに対してリードを疑似整列させ、その過程で転写産物の発現を定量化し、 、転写産物レベルの発現を遺伝子レベルの発現に要約する。 この方法の利点は、計算の効率化、DNA汚染の影響の緩和、GCバイアスの補正などである。 例:
salmon quant --gcBias+tximport
一般的なシーケンスリード深度では、遺伝子発現定量は転写産物発現定量よりも正確であることが多い。 しかし、遺伝子発現の差分解析は、転写産物レベルの定量化にもアクセスすることで、改善 。
RNA-seqの定量に使われる他のツールには以下のようなものがある:TopHat2、bowtie2、kallisto、HTseqなどである。
適切なRNA-seq定量化の選択は、多くの要因の中でも、トランスクリプトームアノテーションの質、 、RNA-seqライブラリー調製の質、コンタミネーション配列の有無に依存する。 多くの場合、複数のアプローチによる定量結果を比較することは有益である。
最適な定量化方法は生物種や実験に依存し、しばしば大量の計算資源を必要とするため、このワークショップではカウントの生成方法に関する具体的な説明は行わない。 その代わりに、上記の参考文献をチェックし、助けが必要であれば地元のバイオインフォマティクスの専門家に相談することをお勧めする。
チャレンジ隣人と以下の点について話し合ってください。
- RNA-Seq定量化ツールについて聞いたことがあるものはどれですか? その他の方法の長所と短所をご存知ですか?
- 独自のRNA-Seq実験を行ったことがありますか? もしそうなら、どのような定量化ツールを使い、なぜそれを選んだのか?
- 定量化のための特定のツール/地元のバイオインフォマティクスの専門家/計算リソースにアクセスできるか? そうでなければ、どうやってアクセスするのか?
参照配列の検索
RNA-seqデータから既知の遺伝子や転写産物の量を定量するためには、これらの特徴の配列を知らせてくれる参照データベースが必要であり、これと我々のリードを比較することができる。 この情報は、さまざまなオンラインレポジトリから得ることができる。 特定のプロジェクトには、異なるソースからの情報を混在させず、これらのうちの1つを選択することを強く推奨する。 選択した定量化ツールによって、必要な参照情報の種類は異なる。 リードをゲノムにアライメントし、既知の注釈付きフィーチャーとのオーバーラップを調べる場合、全ゲノム配列(fastaファイルで提供)と、各注釈付きフィーチャーのゲノム上の位置を示すファイル(通常gtfファイルで提供)が必要です。 リードをトランスクリプトームにマッピングする場合は、代わりに各トランスクリプトの配列を含むファイル(これもfastaファイル)が必要になります。
- マウスやヒトのサンプルを扱う場合、GENCODEプロジェクトは、十分にキュレートされた参照ファイルを提供している。
- Ensemblは、植物や菌類を含む、多数の生物の参照ファイルを提供している。
- また、UCSCは、多くの生物の参照ファイルを提供している。
チャレンジ
GENCODEから最新のマウストランスクリプトームファスタファイルをダウンロードする。
エントリーはどのようなものですか?
ヒント:ファイルをRに読み込むには、Biostrings パッケージの
readDNAStringSet() 関数を使う。
このワークショップで我々はどこに向かっているのか?
この2日間で、Bioconductorを使ってどのように微分発現解析を行うか、そして結果をどのように解釈するかについて議論し、実践する。 カウントマトリックスから始めるので、最初の品質評価と遺伝子発現の定量はすでに行われていると仮定する。 差次的発現解析の結果は、MAプロットやヒートマップなどのグラフを用いて表現されることが多い(例は下記参照)。
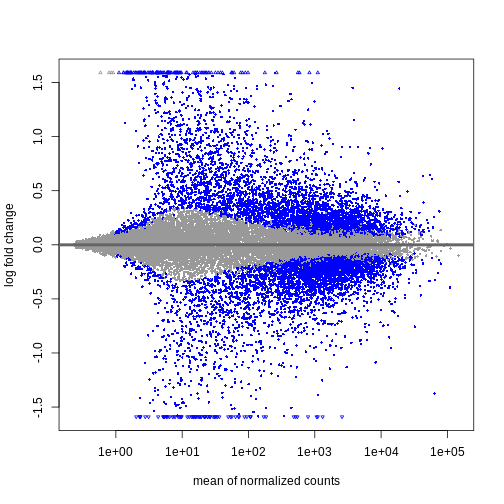
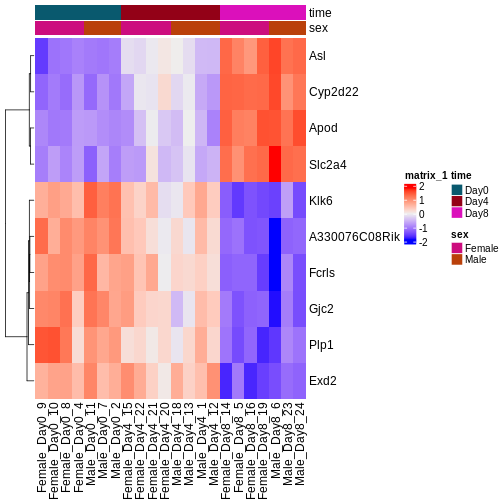
以下のエピソードでは、特にこれらのプロットの作成と解釈の仕方を学ぶ。 また、上位にランクされた遺伝子間に機能的関係があるかどうかを調べるために、遺伝子セット(濃縮)解析と呼ばれるフォローアップ解析を行うことも一般的であるが、これについても後のエピソードで取り上げる。
- RNA-seqは、ある細胞/組織内で発現しているRNAの量と、ある時点での状態を測定する技術である。
- RNA-seq実験を計画する際には、ポリA選択とリボソーム除去のどちらを行うか、ストランドプロトコルとアンストランドプロトコルのどちらを適用するか、シングルエンドとペアエンドのどちらでリードをシーケンスするかなど、多くの選択をしなければならない。 それぞれの選択は、データの処理と解釈に結果をもたらす。
- RNA-seqデータの定量化には多くのアプローチが存在する。 リードをゲノムにアライメントし、遺伝子座にオーバーラップするリードの数をカウントする方法もある。 他の方法は、リードをトランスクリプトームにマッピングし、確率的アプローチを使って各遺伝子や転写産物の存在量を推定する。
- 注釈付き遺伝子に関する情報は、Ensembl、UCSC、GENCODEなどいくつかの情報源からアクセスできる。