dplyrによるデータの操作と分析
Last updated on 2026-04-28 | Edit this page
Overview
Questions
- tidyverseメタパッケージを使用したRによるデータ分析
Objectives
-
dplyrパッケージとtidyrパッケージの目的について説明してください。 - データを操作する際に特に有用な主要な関数をいくつか挙げてください。
- ワイド形式とロング形式のテーブル概念について説明し、データフレームを一方の形式から他方の形式に変換する方法を示してください。
- テーブルの結合方法を具体的にデモンストレーションしてください。
本エピソードは、Data Carpentriesの「生態学者向けRによるデータ分析と可視化」講座を基にしています。
dplyrとtidyrを用いたデータ操作
ブラケットサブセットは便利ですが、複雑な操作を行う場合、煩雑で可読性が低下することがあります。
データ操作を行う際、特定のパッケージを活用することで作業を大幅に効率化できます。Rにおけるパッケージとは、基本的に追加機能を提供する関数群であり、これまで使用してきたstr()やdata.frame()などの関数はRに標準で組み込まれています。一方、パッケージを導入することで、さらに専門的な機能を利用できるようになります。初めてパッケージを使用する前には必ずインストールが必要で、その後は必要に応じて各Rセッションでインポートする必要があります。
dplyrパッケージは強力なデータ操作ツールを提供します。データフレームを直接操作するために設計されており、多くの操作が最適化されています。後述するように、特定の分析や可視化を行うためにデータフレームの形状を変更したい場合があります。
tidyrパッケージはこの一般的な問題に対処するためのツールを提供し、データをきれいに整理した状態で操作できるようにします。
ワークショップ終了後、dplyrとtidyrについてさらに詳しく学びたい場合は、以下の資料が参考になります:
- dplyrを使ったデータ変換の便利なチートシート
- tidyrに関するチートシート
-
tidyverseパッケージは「包括的パッケージ」であり、tidyr、dplyr、ggplot2、tibbleなど、データ分析において相互に連携して動作する複数の有用なパッケージをまとめてインストールできます。これらのパッケージはデータの操作や分析を容易にし、サブセット処理、データ変換、可視化など、さまざまな操作を可能にします。
セットアップを完了していれば、すでにtidyverseパッケージがインストールされているはずです。以下のコマンドでライブラリから読み込んで確認できます:
R
## tidyverseパッケージ(dplyrを含む)を読み込む
library("tidyverse")
もしthere is no package called ‘tidyverse’というエラーメッセージが表示された場合、このRバージョンに対してまだパッケージがインストールされていません。tidyverseパッケージをインストールするには、以下のコマンドを実行してください:
R
BiocManager::install("tidyverse")
tidyverseパッケージをインストールした場合は、このRセッションでlibrary()コマンドを使用して必ず読み込んでください!
tidyverseを使ったデータ読み込み
read.csv()の代わりに、tidyverseパッケージreadrに含まれるread_csv()関数(.ではなく_を使用している点に注意)を使ってデータを読み込みます。
R
rna <- read_csv("data/rnaseq.csv")
## データの内容を確認
rna
OUTPUT
# A tibble: 32,428 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>データのクラスが”tibble”として表示されていることにご注意ください。
tibbleは、以前紹介したデータフレームオブジェクトの動作を一部調整したものです。データ構造はデータフレームと非常に似ていますが、主な違いは以下の通りです:
各列のデータ型が列名の下に表示されます。 注:<
dbl>は小数点を含む数値値を保持するように定義されたデータ型です。データの最初の数行のみが表示され、1画面に収まる列数のみが出力されます。
ここからは、dplyrでよく使われる代表的な関数をいくつか紹介します:
-
select():列のサブセットを選択 -
filter():条件に基づいて行をサブセット -
mutate():他の列の情報を使って新しい列を作成 -
group_by()とsummarise():グループ化されたデータの要約統計量を作成 -
arrange():結果を並べ替え -
count():離散値の出現回数をカウント
列の選択と行のフィルタリング
データフレームから特定の列を選択するには select()
関数を使用します。この関数の第1引数には対象のデータフレーム(ここでは
rna)を指定し、続く引数には保持したい列名を指定します。
R
select(rna, gene, sample, tissue, expression)
OUTPUT
# A tibble: 32,428 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545336 Cerebellum 1170
2 Apod GSM2545336 Cerebellum 36194
3 Cyp2d22 GSM2545336 Cerebellum 4060
4 Klk6 GSM2545336 Cerebellum 287
5 Fcrls GSM2545336 Cerebellum 85
6 Slc2a4 GSM2545336 Cerebellum 782
7 Exd2 GSM2545336 Cerebellum 1619
8 Gjc2 GSM2545336 Cerebellum 288
9 Plp1 GSM2545336 Cerebellum 43217
10 Gnb4 GSM2545336 Cerebellum 1071
# ℹ 32,418 more rows特定の列を除いたすべての列を選択する場合は、除外したい列名の前に「-」を付けます。
R
select(rna, -tissue, -organism)
OUTPUT
# A tibble: 32,428 × 17
gene sample expression age sex infection strain time mouse ENTREZID
<chr> <chr> <dbl> <dbl> <chr> <chr> <chr> <dbl> <dbl> <dbl>
1 Asl GSM2545… 1170 8 Fema… Influenz… C57BL… 8 14 109900
2 Apod GSM2545… 36194 8 Fema… Influenz… C57BL… 8 14 11815
3 Cyp2d22 GSM2545… 4060 8 Fema… Influenz… C57BL… 8 14 56448
4 Klk6 GSM2545… 287 8 Fema… Influenz… C57BL… 8 14 19144
5 Fcrls GSM2545… 85 8 Fema… Influenz… C57BL… 8 14 80891
6 Slc2a4 GSM2545… 782 8 Fema… Influenz… C57BL… 8 14 20528
7 Exd2 GSM2545… 1619 8 Fema… Influenz… C57BL… 8 14 97827
8 Gjc2 GSM2545… 288 8 Fema… Influenz… C57BL… 8 14 118454
9 Plp1 GSM2545… 43217 8 Fema… Influenz… C57BL… 8 14 18823
10 Gnb4 GSM2545… 1071 8 Fema… Influenz… C57BL… 8 14 14696
# ℹ 32,418 more rows
# ℹ 7 more variables: product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>これにより、rna データフレームから tissue
列と organism 列を除いたすべての変数が選択されます。
特定の条件に基づいて行を選択する場合は filter()
関数を使用します:
R
filter(rna, sex == "Male")
OUTPUT
# A tibble: 14,740 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 626 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
2 Apod GSM254… 13021 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
3 Cyp2d22 GSM254… 2171 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
4 Klk6 GSM254… 448 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
5 Fcrls GSM254… 180 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
6 Slc2a4 GSM254… 313 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
7 Exd2 GSM254… 2366 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
8 Gjc2 GSM254… 310 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
9 Plp1 GSM254… 53126 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
10 Gnb4 GSM254… 1355 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
# ℹ 14,730 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>R
filter(rna, sex == "Male" & infection == "NonInfected")
OUTPUT
# A tibble: 4,422 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 535 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
2 Apod GSM254… 13668 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
3 Cyp2d22 GSM254… 2008 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
4 Klk6 GSM254… 1101 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
5 Fcrls GSM254… 375 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
6 Slc2a4 GSM254… 249 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
7 Exd2 GSM254… 3126 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
8 Gjc2 GSM254… 791 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
9 Plp1 GSM254… 98658 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
10 Gnb4 GSM254… 2437 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
# ℹ 4,412 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>次に、このデータセットで解析されたマウス遺伝子のヒト相同遺伝子について調べたいとします。この情報は
rna データフレームの最後の列
hsapiens_homolog_associated_gene_name
に格納されています。この情報を視覚化しやすくするため、gene
列と hsapiens_homolog_associated_gene_name
列のみを含む新しいテーブルを作成します。
R
genes <- select(rna, gene, hsapiens_homolog_associated_gene_name)
genes
OUTPUT
# A tibble: 32,428 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Asl ASL
2 Apod APOD
3 Cyp2d22 CYP2D6
4 Klk6 KLK6
5 Fcrls FCRL2
6 Slc2a4 SLC2A4
7 Exd2 EXD2
8 Gjc2 GJC2
9 Plp1 PLP1
10 Gnb4 GNB4
# ℹ 32,418 more rowsマウス遺伝子の中にはヒト相同遺伝子が存在しないものもあります。これらの遺伝子は
filter() 関数と is.na()
関数を使用して取得できます。is.na() 関数は値が
NA かどうかを判定します。
R
filter(genes, is.na(hsapiens_homolog_associated_gene_name))
OUTPUT
# A tibble: 4,290 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Prodh <NA>
2 Tssk5 <NA>
3 Vmn2r1 <NA>
4 Gm10654 <NA>
5 Hexa <NA>
6 Sult1a1 <NA>
7 Gm6277 <NA>
8 Tmem198b <NA>
9 Adam1a <NA>
10 Ebp <NA>
# ℹ 4,280 more rowsヒト相同遺伝子が存在するマウス遺伝子のみを保持したい場合は、結果を否定する
!
記号を使用します。これにより、hsapiens_homolog_associated_gene_name
が NA でないすべての行を選択できます。
R
filter(genes, !is.na(hsapiens_homolog_associated_gene_name))
OUTPUT
# A tibble: 28,138 × 2
gene hsapiens_homolog_associated_gene_name
<chr> <chr>
1 Asl ASL
2 Apod APOD
3 Cyp2d22 CYP2D6
4 Klk6 KLK6
5 Fcrls FCRL2
6 Slc2a4 SLC2A4
7 Exd2 EXD2
8 Gjc2 GJC2
9 Plp1 PLP1
10 Gnb4 GNB4
# ℹ 28,128 more rowsパイプ演算子
列選択とフィルタリングを同時に行いたい場合、以下の3つの方法があります:中間ステップを使用する方法、関数をネストする方法、またはパイプ演算子を使用する方法です。
中間ステップを使用する場合、一時的なデータフレームを作成してそれを次の関数の入力として渡します。例えば:
R
rna2 <- filter(rna, sex == "Male")
rna3 <- select(rna2, gene, sample, tissue, expression)
rna3
OUTPUT
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsこの方法は可読性は高いですが、中間オブジェクトが多数生成されるため、ワークスペースが乱雑になりがちです。特に複数のステップがある場合、管理が難しくなることがあります。
関数をネストする方法もあります:
R
rna3 <- select(filter(rna, sex == "Male"), gene, sample, tissue, expression)
rna3
OUTPUT
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsこの方法は便利ですが、関数がネストしすぎると読みにくくなる可能性があります。Rは式を内側から評価するため(この場合、まずフィルタリングを行い、その後選択を行います)、複雑な処理になると理解が難しくなる場合があります。
最後の選択肢である「パイプ演算子」はRに最近追加された機能です。パイプ演算子を使用すると、ある関数の出力を直接次の関数の入力として渡すことができ、同じデータセットに対して複数の処理を行う場合に特に便利です。
Rでパイプ演算子を使用するには %>%
記号を使用します(magrittr
パッケージで利用可能)。また |>
記号も使用できます(基本Rで利用可能)。RStudioを使用している場合、PCではCtrl
+ Shift + M、MacではCmd +
Shift + M でパイプ演算子を入力できます。
上記のコードでは、パイプ演算子を使用して rna
データセットを最初に filter() 関数で sex が
“Male” の行のみを保持するようにフィルタリングし、次に
select() 関数で gene, sample,
tissue, expression
列のみを保持するように選択しています。
|>
パイプ演算子は左側のオブジェクトを右側の関数の第1引数として直接渡すため、filter()
や select()
関数にデータフレームを明示的に引数として指定する必要がなくなります。
R
rna |>
filter(sex == "Male") |>
select(gene, sample, tissue, expression)
OUTPUT
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rowsパイプ演算子を「その後」という意味で読むと理解しやすいかもしれません。例えば上記の例では、rna
データフレームを最初に取得し、その後 sex == "Male"
の行をフィルタリングし、その後 gene,
sample, tissue, expression
列を選択しています。
dplyr
関数自体はシンプルな機能ですが、パイプ演算子を使ってこれらの関数を線形的なワークフローに組み合わせることで、より複雑なデータフレーム操作が可能になります。
この小さなサイズのデータセットから新しいオブジェクトを作成したい場合は、以下のように新しい名前を割り当てられます:
R
rna3 <- rna |>
filter(sex == "Male") |>
select(gene, sample, tissue, expression)
rna3
OUTPUT
# A tibble: 14,740 × 4
gene sample tissue expression
<chr> <chr> <chr> <dbl>
1 Asl GSM2545340 Cerebellum 626
2 Apod GSM2545340 Cerebellum 13021
3 Cyp2d22 GSM2545340 Cerebellum 2171
4 Klk6 GSM2545340 Cerebellum 448
5 Fcrls GSM2545340 Cerebellum 180
6 Slc2a4 GSM2545340 Cerebellum 313
7 Exd2 GSM2545340 Cerebellum 2366
8 Gjc2 GSM2545340 Cerebellum 310
9 Plp1 GSM2545340 Cerebellum 53126
10 Gnb4 GSM2545340 Cerebellum 1355
# ℹ 14,730 more rows課題:
パイプ演算子を使用して、rnaデータセットをサブセットし、時間0時点で雌マウスの観測値のみを保持してください。
この際、遺伝子の発現量が50,000を超えているものに限定し、列はgene、sample、time、expression、ageの5列のみを抽出してください。
解答
R
rna |>
filter(expression > 50000,
sex == "Female",
time == 0 ) |>
select(gene, sample, time, expression, age)
OUTPUT
# A tibble: 9 × 5
gene sample time expression age
<chr> <chr> <dbl> <dbl> <dbl>
1 Plp1 GSM2545337 0 101241 8
2 Atp1b1 GSM2545337 0 53260 8
3 Plp1 GSM2545338 0 96534 8
4 Atp1b1 GSM2545338 0 50614 8
5 Plp1 GSM2545348 0 102790 8
6 Atp1b1 GSM2545348 0 59544 8
7 Plp1 GSM2545353 0 71237 8
8 Glul GSM2545353 0 52451 8
9 Atp1b1 GSM2545353 0 61451 8mutate関数の使用
既存の列の値に基づいて新しい列を作成したい場面は頻繁に発生します。例えば、単位変換を行ったり、2つの列間の値の比率を計算したりする場合などです。このような場合には、mutate()関数を使用します。
時間の値を時間単位の新しい列として作成する例を示します:
R
rna |>
mutate(time_hours = time * 24) |>
select(time, time_hours)
OUTPUT
# A tibble: 32,428 × 2
time time_hours
<dbl> <dbl>
1 8 192
2 8 192
3 8 192
4 8 192
5 8 192
6 8 192
7 8 192
8 8 192
9 8 192
10 8 192
# ℹ 32,418 more rowsまた、mutate()関数の同じ呼び出し内で、先に作成した新しい列を基にさらに別の新しい列を作成することも可能です:
R
rna |>
mutate(time_hours = time * 24,
time_mn = time_hours * 60) |>
select(time, time_hours, time_mn)
OUTPUT
# A tibble: 32,428 × 3
time time_hours time_mn
<dbl> <dbl> <dbl>
1 8 192 11520
2 8 192 11520
3 8 192 11520
4 8 192 11520
5 8 192 11520
6 8 192 11520
7 8 192 11520
8 8 192 11520
9 8 192 11520
10 8 192 11520
# ℹ 32,418 more rows課題
rna
データセットから、以下の条件を満たす新しいデータフレームを作成してください:
gene、chromosome_name、phenotype_description、sample、および
expression
の各列のみを含むこと。発現値については対数変換を施すこと。このデータフレームには、性染色体上に位置し、特定の表現型_descriptionに関連付けられ、かつ対数変換後の発現値が5を超える遺伝子のみを含める必要があります。
ヒント:このデータフレームを生成するために、コマンドをどのように順序付ければよいか考えてみてください!
R
rna |>
mutate(expression = log(expression)) |>
select(gene, chromosome_name, phenotype_description, sample, expression) |>
filter(chromosome_name == "X" | chromosome_name == "Y") |>
filter(!is.na(phenotype_description)) |>
filter(expression > 5)
OUTPUT
# A tibble: 649 × 5
gene chromosome_name phenotype_description sample expression
<chr> <chr> <chr> <chr> <dbl>
1 Plp1 X abnormal CNS glial cell morphology GSM25… 10.7
2 Slc7a3 X decreased body length GSM25… 5.46
3 Plxnb3 X abnormal coat appearance GSM25… 6.58
4 Rbm3 X abnormal liver morphology GSM25… 9.32
5 Cfp X abnormal cardiovascular system phys… GSM25… 6.18
6 Ebp X abnormal embryonic erythrocyte morp… GSM25… 6.68
7 Cd99l2 X abnormal cellular extravasation GSM25… 8.04
8 Piga X abnormal brain development GSM25… 6.06
9 Pim2 X decreased T cell proliferation GSM25… 7.11
10 Itm2a X no abnormal phenotype detected GSM25… 7.48
# ℹ 639 more rowsSplit-apply-combine data analysis
Many data analysis tasks can be approached using the
split-apply-combine paradigm: split the data into groups, apply
some analysis to each group, and then combine the results.
dplyr makes this very easy through the use
of the group_by() function.
R
rna |>
group_by(gene)
OUTPUT
# A tibble: 32,428 × 19
# Groups: gene [1,474]
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>The group_by() function doesn’t perform any data
processing, it groups the data into subsets: in the example above, our
initial tibble of 32428 observations is split into 1474
groups based on the gene variable.
We could similarly decide to group the tibble by the samples:
R
rna |>
group_by(sample)
OUTPUT
# A tibble: 32,428 × 19
# Groups: sample [22]
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 Asl GSM254… 1170 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 Apod GSM254… 36194 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
3 Cyp2d22 GSM254… 4060 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
4 Klk6 GSM254… 287 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
5 Fcrls GSM254… 85 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
6 Slc2a4 GSM254… 782 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
7 Exd2 GSM254… 1619 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 Gjc2 GSM254… 288 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
9 Plp1 GSM254… 43217 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
10 Gnb4 GSM254… 1071 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>Here our initial tibble of 32428 observations is split
into 22 groups based on the sample variable.
Once the data has been grouped, subsequent operations will be applied on each group independently.
The summarise() function
group_by() is often used together with
summarise(), which collapses each group into a single-row
summary of that group.
group_by() takes as arguments the column names that
contain the categorical variables for which you want to
calculate the summary statistics. So to compute the mean
expression by gene:
R
rna |>
group_by(gene) |>
summarise(mean_expression = mean(expression))
OUTPUT
# A tibble: 1,474 × 2
gene mean_expression
<chr> <dbl>
1 AI504432 1053.
2 AW046200 131.
3 AW551984 295.
4 Aamp 4751.
5 Abca12 4.55
6 Abcc8 2498.
7 Abhd14a 525.
8 Abi2 4909.
9 Abi3bp 1002.
10 Abl2 2124.
# ℹ 1,464 more rowsWe could also want to calculate the mean expression levels of all genes in each sample:
R
rna |>
group_by(sample) |>
summarise(mean_expression = mean(expression))
OUTPUT
# A tibble: 22 × 2
sample mean_expression
<chr> <dbl>
1 GSM2545336 2062.
2 GSM2545337 1766.
3 GSM2545338 1668.
4 GSM2545339 1696.
5 GSM2545340 1682.
6 GSM2545341 1638.
7 GSM2545342 1594.
8 GSM2545343 2107.
9 GSM2545344 1712.
10 GSM2545345 1700.
# ℹ 12 more rowsBut we can can also group by multiple columns:
R
rna |>
group_by(gene, infection, time) |>
summarise(mean_expression = mean(expression))
OUTPUT
`summarise()` has grouped output by 'gene', 'infection'. You can override using
the `.groups` argument.OUTPUT
# A tibble: 4,422 × 4
# Groups: gene, infection [2,948]
gene infection time mean_expression
<chr> <chr> <dbl> <dbl>
1 AI504432 InfluenzaA 4 1104.
2 AI504432 InfluenzaA 8 1014
3 AI504432 NonInfected 0 1034.
4 AW046200 InfluenzaA 4 152.
5 AW046200 InfluenzaA 8 81
6 AW046200 NonInfected 0 155.
7 AW551984 InfluenzaA 4 302.
8 AW551984 InfluenzaA 8 342.
9 AW551984 NonInfected 0 238
10 Aamp InfluenzaA 4 4870
# ℹ 4,412 more rowsOnce the data is grouped, you can also summarise multiple variables
at the same time (and not necessarily on the same variable). For
instance, we could add a column indicating the median
expression by gene and by condition:
R
rna |>
group_by(gene, infection, time) |>
summarise(mean_expression = mean(expression),
median_expression = median(expression))
OUTPUT
`summarise()` has grouped output by 'gene', 'infection'. You can override using
the `.groups` argument.OUTPUT
# A tibble: 4,422 × 5
# Groups: gene, infection [2,948]
gene infection time mean_expression median_expression
<chr> <chr> <dbl> <dbl> <dbl>
1 AI504432 InfluenzaA 4 1104. 1094.
2 AI504432 InfluenzaA 8 1014 985
3 AI504432 NonInfected 0 1034. 1016
4 AW046200 InfluenzaA 4 152. 144.
5 AW046200 InfluenzaA 8 81 82
6 AW046200 NonInfected 0 155. 163
7 AW551984 InfluenzaA 4 302. 245
8 AW551984 InfluenzaA 8 342. 287
9 AW551984 NonInfected 0 238 265
10 Aamp InfluenzaA 4 4870 4708
# ℹ 4,412 more rowsチャレンジ
時間ポイントごとに遺伝子「Dok3」の平均発現レベルを計算します。
R
rna |>
filter(gene == "Dok3") |>
group_by(time) |>
summarise(mean = mean(expression))
OUTPUT
# A tibble: 3 × 2
time mean
<dbl> <dbl>
1 0 169
2 4 156.
3 8 61 カウント処理
データを扱う際、各因子または因子の組み合わせごとに観測値の数を知りたい場面がよくあります。この目的のために、dplyr
パッケージでは count()
関数が用意されています。例えば、感染サンプルと非感染サンプルそれぞれについてデータ行数をカウントしたい場合、以下のように記述します:
R
rna |>
count(infection)
OUTPUT
# A tibble: 2 × 2
infection n
<chr> <int>
1 InfluenzaA 22110
2 NonInfected 10318count()
関数は、既に学んだ「変数でデータをグループ化し、そのグループ内の観測値数を集計する」という処理の短縮形です。言い換えれば、rna |> count(infection)
は以下のように等価です:
R
rna |>
group_by(infection) |>
summarise(n = n())
OUTPUT
# A tibble: 2 × 2
infection n
<chr> <int>
1 InfluenzaA 22110
2 NonInfected 10318上記の例では、count() 関数を使って単一の因子(ここでは
infection)の行数をカウントしています。 もし
infection と time
といった複数の因子の組み合わせについてカウントを行いたい場合は、count()
関数の引数として最初の因子と2番目の因子を指定します:
R
rna |>
count(infection, time)
OUTPUT
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318これは以下のコードと等価です:
R
rna |>
group_by(infection, time) |>
summarise(n = n())
OUTPUT
`summarise()` has grouped output by 'infection'. You can override using the
`.groups` argument.OUTPUT
# A tibble: 3 × 3
# Groups: infection [2]
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318結果を並べ替えて比較を容易にすることが役立つ場合があります。
arrange() 関数を使えばテーブルを並べ替えることが可能です。
例えば、先ほどのテーブルを time
で昇順に並べ替えたい場合は以下のようにします:
R
rna |>
count(infection, time) |>
arrange(time)
OUTPUT
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 NonInfected 0 10318
2 InfluenzaA 4 11792
3 InfluenzaA 8 10318あるいは、カウント値で並べ替える場合は:
R
rna |>
count(infection, time) |>
arrange(n)
OUTPUT
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 8 10318
2 NonInfected 0 10318
3 InfluenzaA 4 11792降順で並べ替える場合は、desc()
関数を追加する必要があります:
R
rna |>
count(infection, time) |>
arrange(desc(n))
OUTPUT
# A tibble: 3 × 3
infection time n
<chr> <dbl> <int>
1 InfluenzaA 4 11792
2 InfluenzaA 8 10318
3 NonInfected 0 10318チャレンジ
- 各サンプルにおいて解析された遺伝子数はいくつですか?
-
group_by()とsummarise()関数を使用して、各サンプルにおけるシーケンシング深度(すべてのカウントの合計)を評価してください。最もシーケンシング深度が高いサンプルはどれですか? - 任意の1つのサンプルを選択し、遺伝子をバイオタイプ別に分類して数を評価してください。
- 「異常なDNAメチル化」という表現型記述に関連する遺伝子を特定し、時間0、時間4、および時間8におけるそれらの平均発現量(対数値)を算出してください。
R
## 1.
rna |>
count(sample)
OUTPUT
# A tibble: 22 × 2
sample n
<chr> <int>
1 GSM2545336 1474
2 GSM2545337 1474
3 GSM2545338 1474
4 GSM2545339 1474
5 GSM2545340 1474
6 GSM2545341 1474
7 GSM2545342 1474
8 GSM2545343 1474
9 GSM2545344 1474
10 GSM2545345 1474
# ℹ 12 more rowsR
## 2.
rna |>
group_by(sample) |>
summarise(seq_depth = sum(expression)) |>
arrange(desc(seq_depth))
OUTPUT
# A tibble: 22 × 2
sample seq_depth
<chr> <dbl>
1 GSM2545350 3255566
2 GSM2545352 3216163
3 GSM2545343 3105652
4 GSM2545336 3039671
5 GSM2545380 3036098
6 GSM2545353 2953249
7 GSM2545348 2913678
8 GSM2545362 2913517
9 GSM2545351 2782464
10 GSM2545349 2758006
# ℹ 12 more rowsR
## 3.
rna |>
filter(sample == "GSM2545336") |>
count(gene_biotype) |>
arrange(desc(n))
OUTPUT
# A tibble: 13 × 2
gene_biotype n
<chr> <int>
1 protein_coding 1321
2 lncRNA 69
3 processed_pseudogene 59
4 miRNA 7
5 snoRNA 5
6 TEC 4
7 polymorphic_pseudogene 2
8 unprocessed_pseudogene 2
9 IG_C_gene 1
10 scaRNA 1
11 transcribed_processed_pseudogene 1
12 transcribed_unitary_pseudogene 1
13 transcribed_unprocessed_pseudogene 1R
## 4.
rna |>
filter(phenotype_description == "abnormal DNA methylation") |>
group_by(gene, time) |>
summarise(mean_expression = mean(log(expression))) |>
arrange()
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.OUTPUT
# A tibble: 6 × 3
# Groups: gene [2]
gene time mean_expression
<chr> <dbl> <dbl>
1 Xist 0 6.95
2 Xist 4 6.34
3 Xist 8 7.13
4 Zdbf2 0 6.27
5 Zdbf2 4 6.27
6 Zdbf2 8 6.19データの再整形
rna
データフレームにおいて、行には発現値(単位)が格納されており、これらは
gene と sample
という2つの他の変数の組み合わせに関連付けられています。
その他のすべての列は、サンプルに関する変数(生物種、年齢、性別など)または遺伝子に関する変数 (gene_biotype、ENTREZ_ID、産物情報など)を記述しています。 遺伝子やサンプルによって変化しない変数は、すべての行で同じ値を保持します。
R
rna |>
arrange(gene)
OUTPUT
# A tibble: 32,428 × 19
gene sample expression organism age sex infection strain time tissue
<chr> <chr> <dbl> <chr> <dbl> <chr> <chr> <chr> <dbl> <chr>
1 AI504432 GSM25… 1230 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
2 AI504432 GSM25… 1085 Mus mus… 8 Fema… NonInfec… C57BL… 0 Cereb…
3 AI504432 GSM25… 969 Mus mus… 8 Fema… NonInfec… C57BL… 0 Cereb…
4 AI504432 GSM25… 1284 Mus mus… 8 Fema… Influenz… C57BL… 4 Cereb…
5 AI504432 GSM25… 966 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
6 AI504432 GSM25… 918 Mus mus… 8 Male Influenz… C57BL… 8 Cereb…
7 AI504432 GSM25… 985 Mus mus… 8 Fema… Influenz… C57BL… 8 Cereb…
8 AI504432 GSM25… 972 Mus mus… 8 Male NonInfec… C57BL… 0 Cereb…
9 AI504432 GSM25… 1000 Mus mus… 8 Fema… Influenz… C57BL… 4 Cereb…
10 AI504432 GSM25… 816 Mus mus… 8 Male Influenz… C57BL… 4 Cereb…
# ℹ 32,418 more rows
# ℹ 9 more variables: mouse <dbl>, ENTREZID <dbl>, product <chr>,
# ensembl_gene_id <chr>, external_synonym <chr>, chromosome_name <chr>,
# gene_biotype <chr>, phenotype_description <chr>,
# hsapiens_homolog_associated_gene_name <chr>この構造は「長形式」と呼ばれ、1つの列にすべての値が含まれ、他の列がその値の文脈情報を列挙する形式です。 特定のケースでは、この「長形式」は「人間にとって読みやすい」形式とは言えず、よりコンパクトなデータ表現方法である 「幅形式」が好まれる場合があります。これは特に、科学者が行列形式で見ることに慣れている遺伝子発現値の場合に当てはまります。 この形式では、行が遺伝子を、列がサンプルを表すため、遺伝子発現レベルのサンプル内およびサンプル間の関係性を 容易に探索できるようになります。
OUTPUT
# A tibble: 1,474 × 23
gene GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988
2 Apod 36194 10347 9173 10620 13021 29594
3 Cyp2d22 4060 1616 1603 1901 2171 3349
4 Klk6 287 629 641 578 448 195
5 Fcrls 85 233 244 237 180 38
6 Slc2a4 782 231 248 265 313 786
7 Exd2 1619 2288 2235 2513 2366 1359
8 Gjc2 288 595 568 551 310 146
9 Plp1 43217 101241 96534 58354 53126 27173
10 Gnb4 1071 1791 1867 1430 1355 798
# ℹ 1,464 more rows
# ℹ 16 more variables: GSM2545342 <dbl>, GSM2545343 <dbl>, GSM2545344 <dbl>,
# GSM2545345 <dbl>, GSM2545346 <dbl>, GSM2545347 <dbl>, GSM2545348 <dbl>,
# GSM2545349 <dbl>, GSM2545350 <dbl>, GSM2545351 <dbl>, GSM2545352 <dbl>,
# GSM2545353 <dbl>, GSM2545354 <dbl>, GSM2545362 <dbl>, GSM2545363 <dbl>,
# GSM2545380 <dbl>rna
データの遺伝子発現値を幅形式に変換するには、sample 列の値を
新しいテーブルの列名として使用する新しいテーブルを作成する必要があります。
ここで重要なのは、依然として「tidyデータ構造」の原則に従っているものの、 データの再整形を行っているという点です。つまり、遺伝子ごと・サンプルごとに発現値を記録するのではなく、 遺伝子ごとの発現レベルに焦点を当ててデータを再構成しているのです。
逆の変換としては、列名を新しい変数の値に変換する操作が考えられます。
これら2種類の変換は、tidyr パッケージの
pivot_longer() と pivot_wider()
関数を使用して実行できます (詳細な説明については、tidyr
ウェブサイトのピボット操作に関する 記事を参照してください)。
データをワイド形式に変換する
rnaデータの最初の3列を選択し、pivot_wider()関数を使用してデータをワイド形式に変換します。
R
rna_exp <- rna |>
select(gene, sample, expression)
rna_exp
OUTPUT
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Cyp2d22 GSM2545336 4060
4 Klk6 GSM2545336 287
5 Fcrls GSM2545336 85
6 Slc2a4 GSM2545336 782
7 Exd2 GSM2545336 1619
8 Gjc2 GSM2545336 288
9 Plp1 GSM2545336 43217
10 Gnb4 GSM2545336 1071
# ℹ 32,418 more rowspivot_wider関数には主に3つの引数があります:
- 変換対象のデータ
-
names_from:新しい列名となる値を含む列 -
values_from:新しい列に値を割り当てる元となる列
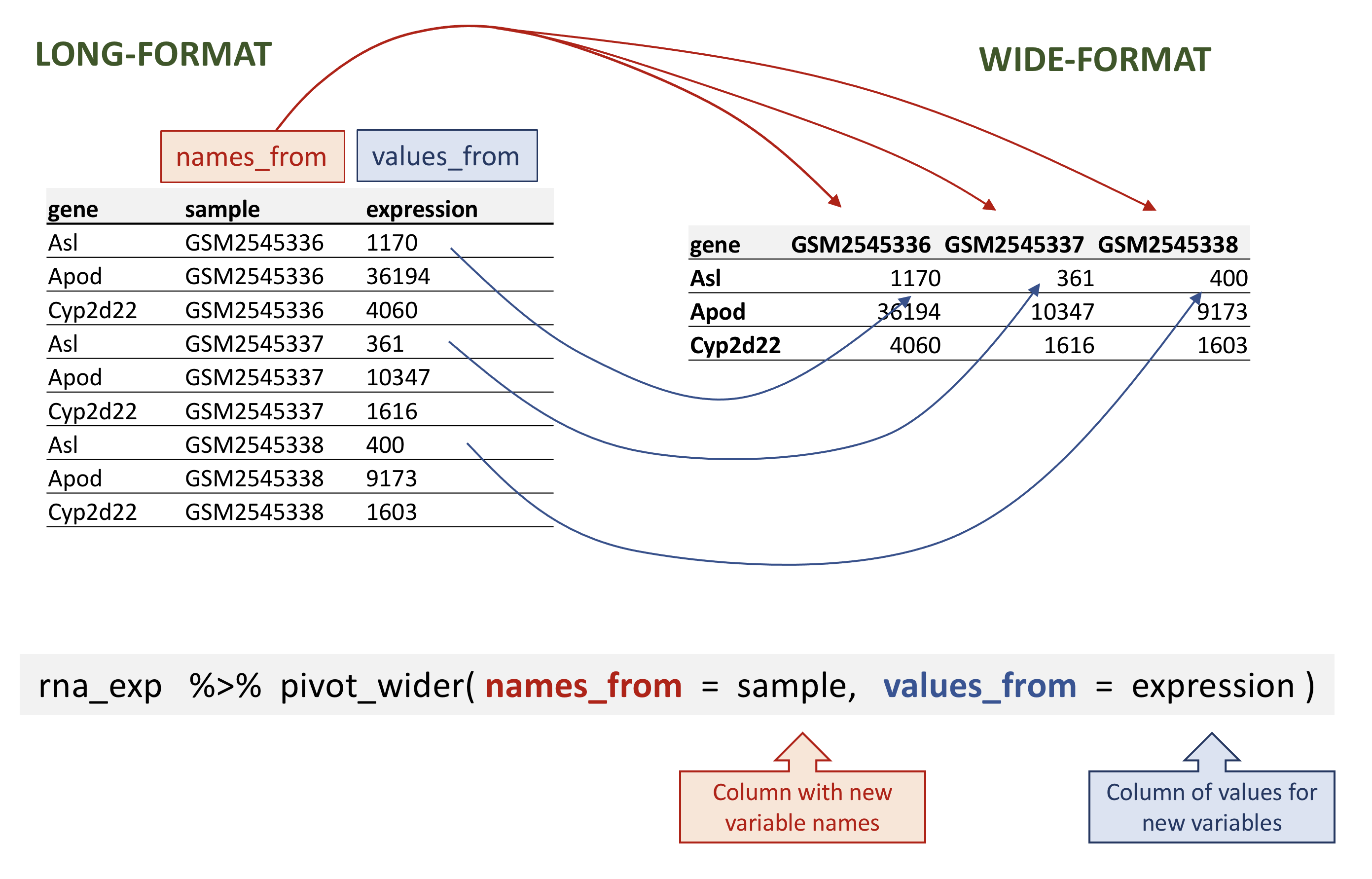
rnaデータのワイド形式への変換例
R
rna_wide <- rna_exp |>
pivot_wider(names_from = sample,
values_from = expression)
rna_wide
OUTPUT
# A tibble: 1,474 × 23
gene GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988
2 Apod 36194 10347 9173 10620 13021 29594
3 Cyp2d22 4060 1616 1603 1901 2171 3349
4 Klk6 287 629 641 578 448 195
5 Fcrls 85 233 244 237 180 38
6 Slc2a4 782 231 248 265 313 786
7 Exd2 1619 2288 2235 2513 2366 1359
8 Gjc2 288 595 568 551 310 146
9 Plp1 43217 101241 96534 58354 53126 27173
10 Gnb4 1071 1791 1867 1430 1355 798
# ℹ 1,464 more rows
# ℹ 16 more variables: GSM2545342 <dbl>, GSM2545343 <dbl>, GSM2545344 <dbl>,
# GSM2545345 <dbl>, GSM2545346 <dbl>, GSM2545347 <dbl>, GSM2545348 <dbl>,
# GSM2545349 <dbl>, GSM2545350 <dbl>, GSM2545351 <dbl>, GSM2545352 <dbl>,
# GSM2545353 <dbl>, GSM2545354 <dbl>, GSM2545362 <dbl>, GSM2545363 <dbl>,
# GSM2545380 <dbl>デフォルトでは、pivot_wider()関数は欠損値がある場合にNA値を追加します。
何らかの理由で、特定の遺伝子について特定のサンプルに発現値の欠損が生じていると仮定しましょう。以下の架空の例では、Cyp2d22遺伝子はGSM2545338サンプルにのみ発現値を持っています。
R
rna_with_missing_values <- rna |>
select(gene, sample, expression) |>
filter(gene %in% c("Asl", "Apod", "Cyp2d22")) |>
filter(sample %in% c("GSM2545336", "GSM2545337", "GSM2545338")) |>
arrange(sample) |>
filter(!(gene == "Cyp2d22" & sample != "GSM2545338"))
rna_with_missing_values
OUTPUT
# A tibble: 7 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Asl GSM2545337 361
4 Apod GSM2545337 10347
5 Asl GSM2545338 400
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545338 1603デフォルトでは、pivot_wider()関数は欠損値がある場合にNA値を追加します。この動作はpivot_wider()関数のvalues_fill引数で変更可能です。
R
rna_with_missing_values |>
pivot_wider(names_from = sample,
values_from = expression)
OUTPUT
# A tibble: 3 × 4
gene GSM2545336 GSM2545337 GSM2545338
<chr> <dbl> <dbl> <dbl>
1 Asl 1170 361 400
2 Apod 36194 10347 9173
3 Cyp2d22 NA NA 1603R
rna_with_missing_values |>
pivot_wider(names_from = sample,
values_from = expression,
values_fill = 0)
OUTPUT
# A tibble: 3 × 4
gene GSM2545336 GSM2545337 GSM2545338
<chr> <dbl> <dbl> <dbl>
1 Asl 1170 361 400
2 Apod 36194 10347 9173
3 Cyp2d22 0 0 1603データを縦長形式に変換する
逆のケースでは、列名を利用してそれらを 2つの新しい変数に変換します。1つ目の変数は列名を値として保持し、 2つ目の変数には従来その列名に関連付けられていた値が格納されます。
pivot_longer() 関数には主に4つの引数があります:
- 変換対象のデータ
-
names_to:作成したい新しい列名で、ここに現在の列名を代入します -
values_to:作成したい新しい列名で、ここに現在の値を代入します -
names_toとvalues_to変数に値を代入する列名(または削除する列名)

rnaデータの縦長変換例
rna_wideデータからrna_longデータを再現する場合、
sampleというキー変数とexpressionという値変数を作成し、
gene列を除くすべての列をキー変数として使用します。ここではgene列を
マイナス記号で削除しています。
新しい変数名を指定する際には引用符で囲む必要がある点に注意してください。
R
rna_long <- rna_wide |>
pivot_longer(names_to = "sample",
values_to = "expression",
-gene)
rna_long
OUTPUT
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Asl GSM2545339 586
5 Asl GSM2545340 626
6 Asl GSM2545341 988
7 Asl GSM2545342 836
8 Asl GSM2545343 535
9 Asl GSM2545344 586
10 Asl GSM2545345 597
# ℹ 32,418 more rows特定の列を指定する方法も使用できます。識別用の列が多数ある場合、
保持する列を指定する方が、除外する列を指定するよりも直感的です。
ここではstarts_with()関数を使用することで、サンプル名をすべて列挙せずに取得できます!
別の方法として、:演算子を使用することもできます!
R
rna_wide |>
pivot_longer(names_to = "sample",
values_to = "expression",
cols = starts_with("GSM"))
OUTPUT
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Asl GSM2545339 586
5 Asl GSM2545340 626
6 Asl GSM2545341 988
7 Asl GSM2545342 836
8 Asl GSM2545343 535
9 Asl GSM2545344 586
10 Asl GSM2545345 597
# ℹ 32,418 more rowsR
rna_wide |>
pivot_longer(names_to = "sample",
values_to = "expression",
GSM2545336:GSM2545380)
OUTPUT
# A tibble: 32,428 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Asl GSM2545339 586
5 Asl GSM2545340 626
6 Asl GSM2545341 988
7 Asl GSM2545342 836
8 Asl GSM2545343 535
9 Asl GSM2545344 586
10 Asl GSM2545345 597
# ℹ 32,418 more rows幅広形式のデータに欠損値が含まれている場合、NA値は縦長形式に変換されたデータにも引き継がれることに注意してください。
以前作成した欠損値を含む仮想的なtibbleを思い出してください:
R
rna_with_missing_values
OUTPUT
# A tibble: 7 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Asl GSM2545337 361
4 Apod GSM2545337 10347
5 Asl GSM2545338 400
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545338 1603R
wide_with_NA <- rna_with_missing_values |>
pivot_wider(names_from = sample,
values_from = expression)
wide_with_NA
OUTPUT
# A tibble: 3 × 4
gene GSM2545336 GSM2545337 GSM2545338
<chr> <dbl> <dbl> <dbl>
1 Asl 1170 361 400
2 Apod 36194 10347 9173
3 Cyp2d22 NA NA 1603R
wide_with_NA |>
pivot_longer(names_to = "sample",
values_to = "expression",
-gene)
OUTPUT
# A tibble: 9 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Asl GSM2545337 361
3 Asl GSM2545338 400
4 Apod GSM2545336 36194
5 Apod GSM2545337 10347
6 Apod GSM2545338 9173
7 Cyp2d22 GSM2545336 NA
8 Cyp2d22 GSM2545337 NA
9 Cyp2d22 GSM2545338 1603データを幅広形式と縦長形式に変換することは、すべての複製サンプルが同じ構成になるようにデータセットを調整する有用な方法です。
質問
マウス遺伝子の中にはヒトにホモログがないものもある。 これらは、
filter() と、 何かが NA かどうかを判定する
is.na() 関数を使って取得することができる。
rnaテーブルから始めて、pivot_wider()関数を使用して、
、各マウスの遺伝子発現レベルを示すワイドフォーマットのテーブルを作成する。
そして、pivot_longer()関数を使って、ロングフォーマットの表を復元する。
R
rna1 <- rna |>
select(gene, mouse, expression) |>
pivot_wider(names_from = mouse, values_from = expression)
rna1
OUTPUT
# A tibble: 1,474 × 23
gene `14` `9` `10` `15` `18` `6` `5` `11` `22` `13` `23`
<chr> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl> <dbl>
1 Asl 1170 361 400 586 626 988 836 535 586 597 938
2 Apod 36194 10347 9173 10620 13021 29594 24959 13668 13230 15868 27769
3 Cyp2d22 4060 1616 1603 1901 2171 3349 3122 2008 2254 2277 2985
4 Klk6 287 629 641 578 448 195 186 1101 537 567 327
5 Fcrls 85 233 244 237 180 38 68 375 199 177 89
6 Slc2a4 782 231 248 265 313 786 528 249 266 357 654
7 Exd2 1619 2288 2235 2513 2366 1359 1474 3126 2379 2173 1531
8 Gjc2 288 595 568 551 310 146 186 791 454 370 240
9 Plp1 43217 101241 96534 58354 53126 27173 28728 98658 61356 61647 38019
10 Gnb4 1071 1791 1867 1430 1355 798 806 2437 1394 1554 960
# ℹ 1,464 more rows
# ℹ 11 more variables: `24` <dbl>, `8` <dbl>, `7` <dbl>, `1` <dbl>, `16` <dbl>,
# `21` <dbl>, `4` <dbl>, `2` <dbl>, `20` <dbl>, `12` <dbl>, `19` <dbl>R
rna1 |>
pivot_longer(names_to = "mouse_id", values_to = "counts", -gene)
OUTPUT
# A tibble: 32,428 × 3
gene mouse_id counts
<chr> <chr> <dbl>
1 Asl 14 1170
2 Asl 9 361
3 Asl 10 400
4 Asl 15 586
5 Asl 18 626
6 Asl 6 988
7 Asl 5 836
8 Asl 11 535
9 Asl 22 586
10 Asl 13 597
# ℹ 32,418 more rows問題
rna
データフレームから、X染色体とY染色体上に位置する遺伝子サブセットを抽出し、
sex を列、chromosome_name
を行、各染色体上の遺伝子の平均発現量を値とした
データフレームを再構成してください。結果は以下の tibble
形式で表示します:
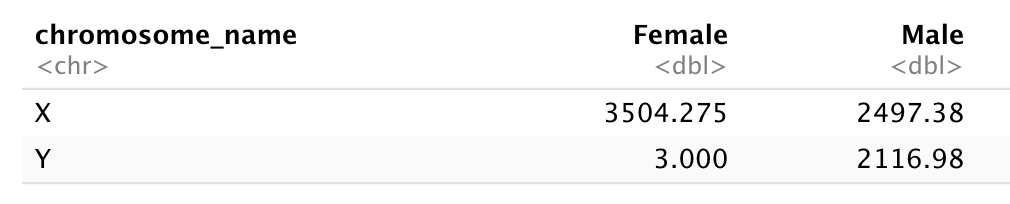
再構成作業の前には必ず要約統計量の計算を行ってください!
まず、男性および女性サンプルからX染色体およびY染色体上の遺伝子の平均発現量を計算します…
R
rna |>
filter(chromosome_name == "Y" | chromosome_name == "X") |>
group_by(sex, chromosome_name) |>
summarise(mean = mean(expression))
OUTPUT
`summarise()` has grouped output by 'sex'. You can override using the `.groups`
argument.OUTPUT
# A tibble: 4 × 3
# Groups: sex [2]
sex chromosome_name mean
<chr> <chr> <dbl>
1 Female X 3504.
2 Female Y 3
3 Male X 2497.
4 Male Y 2117.次に、このデータをワイド形式に変換します
R
rna_1 <- rna |>
filter(chromosome_name == "Y" | chromosome_name == "X") |>
group_by(sex, chromosome_name) |>
summarise(mean = mean(expression)) |>
pivot_wider(names_from = sex,
values_from = mean)
OUTPUT
`summarise()` has grouped output by 'sex'. You can override using the `.groups`
argument.R
rna_1
OUTPUT
# A tibble: 2 × 3
chromosome_name Female Male
<chr> <dbl> <dbl>
1 X 3504. 2497.
2 Y 3 2117.このデータフレームをpivot_longer()関数で変換し、各行が性別と染色体名のユニークな組み合わせに対応するようにします。
R
rna_1 |>
pivot_longer(names_to = "gender",
values_to = "mean",
-chromosome_name)
OUTPUT
# A tibble: 4 × 3
chromosome_name gender mean
<chr> <chr> <dbl>
1 X Female 3504.
2 X Male 2497.
3 Y Female 3
4 Y Male 2117.問題
rna
データセットを使用して、各行が遺伝子の平均発現量を、各列が異なる時間ポイントを表す発現量行列を作成してください。
まず、遺伝子ごとおよび時間ポイントごとに平均発現量を計算しましょう
R
rna |>
group_by(gene, time) |>
summarise(mean_exp = mean(expression))
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.OUTPUT
# A tibble: 4,422 × 3
# Groups: gene [1,474]
gene time mean_exp
<chr> <dbl> <dbl>
1 AI504432 0 1034.
2 AI504432 4 1104.
3 AI504432 8 1014
4 AW046200 0 155.
5 AW046200 4 152.
6 AW046200 8 81
7 AW551984 0 238
8 AW551984 4 302.
9 AW551984 8 342.
10 Aamp 0 4603.
# ℹ 4,412 more rows次に、pivot_wider() 関数を適用します
R
rna_time <- rna |>
group_by(gene, time) |>
summarise(mean_exp = mean(expression)) |>
pivot_wider(names_from = time,
values_from = mean_exp)
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.R
rna_time
OUTPUT
# A tibble: 1,474 × 4
# Groups: gene [1,474]
gene `0` `4` `8`
<chr> <dbl> <dbl> <dbl>
1 AI504432 1034. 1104. 1014
2 AW046200 155. 152. 81
3 AW551984 238 302. 342.
4 Aamp 4603. 4870 4763.
5 Abca12 5.29 4.25 4.14
6 Abcc8 2576. 2609. 2292.
7 Abhd14a 591. 547. 432.
8 Abi2 4881. 4903. 4945.
9 Abi3bp 1175. 1061. 762.
10 Abl2 2170. 2078. 2131.
# ℹ 1,464 more rowsこの操作を行うと、一部の列名が数字で始まるtibbleが生成されます。 時間ポイントに対応する列を選択したい場合、列名を直接指定することはできません… 列4を選択しようとするとどうなるでしょうか?
R
rna |>
group_by(gene, time) |>
summarise(mean_exp = mean(expression)) |>
pivot_wider(names_from = time,
values_from = mean_exp) |>
select(gene, 4)
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.OUTPUT
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene `8`
<chr> <dbl>
1 AI504432 1014
2 AW046200 81
3 AW551984 342.
4 Aamp 4763.
5 Abca12 4.14
6 Abcc8 2292.
7 Abhd14a 432.
8 Abi2 4945.
9 Abi3bp 762.
10 Abl2 2131.
# ℹ 1,464 more rows時間ポイント4を選択するには、列名をバッククォート “`” で囲んで引用符で指定する必要があります
R
rna |>
group_by(gene, time) |>
summarise(mean_exp = mean(expression)) |>
pivot_wider(names_from = time,
values_from = mean_exp) |>
select(gene, `4`)
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.OUTPUT
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene `4`
<chr> <dbl>
1 AI504432 1104.
2 AW046200 152.
3 AW551984 302.
4 Aamp 4870
5 Abca12 4.25
6 Abcc8 2609.
7 Abhd14a 547.
8 Abi2 4903.
9 Abi3bp 1061.
10 Abl2 2078.
# ℹ 1,464 more rows別の方法として、列名を変更し、数字で始まらない名前を選択することも可能です:
R
rna |>
group_by(gene, time) |>
summarise(mean_exp = mean(expression)) |>
pivot_wider(names_from = time,
values_from = mean_exp) |>
rename("time0" = `0`, "time4" = `4`, "time8" = `8`) |>
select(gene, time4)
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.OUTPUT
# A tibble: 1,474 × 2
# Groups: gene [1,474]
gene time4
<chr> <dbl>
1 AI504432 1104.
2 AW046200 152.
3 AW551984 302.
4 Aamp 4870
5 Abca12 4.25
6 Abcc8 2609.
7 Abhd14a 547.
8 Abi2 4903.
9 Abi3bp 1061.
10 Abl2 2078.
# ℹ 1,464 more rows質問
タイムポイントごとの平均発現レベルを含む前のデータフレームを使用し、 、タイムポイント8とタイムポイント0の間のfold-changes、およびタイムポイント8とタイムポイント4の間のfold-changes を含む新しい列を作成する。 この表を、計算されたフォールド・チェンジを集めたロングフォーマットの表に変換する。 この表を、計算されたフォールド・チェンジを集めたロングフォーマットの表に変換する。
rna_time tibbleから開始する:
R
rna_time
OUTPUT
# A tibble: 1,474 × 4
# Groups: gene [1,474]
gene `0` `4` `8`
<chr> <dbl> <dbl> <dbl>
1 AI504432 1034. 1104. 1014
2 AW046200 155. 152. 81
3 AW551984 238 302. 342.
4 Aamp 4603. 4870 4763.
5 Abca12 5.29 4.25 4.14
6 Abcc8 2576. 2609. 2292.
7 Abhd14a 591. 547. 432.
8 Abi2 4881. 4903. 4945.
9 Abi3bp 1175. 1061. 762.
10 Abl2 2170. 2078. 2131.
# ℹ 1,464 more rowsフォールドチェンジを計算する:
R
rna_time |>
mutate(time_8_vs_0 = `8` / `0`, time_8_vs_4 = `8` / `4`)
OUTPUT
# A tibble: 1,474 × 6
# Groups: gene [1,474]
gene `0` `4` `8` time_8_vs_0 time_8_vs_4
<chr> <dbl> <dbl> <dbl> <dbl> <dbl>
1 AI504432 1034. 1104. 1014 0.981 0.918
2 AW046200 155. 152. 81 0.522 0.532
3 AW551984 238 302. 342. 1.44 1.13
4 Aamp 4603. 4870 4763. 1.03 0.978
5 Abca12 5.29 4.25 4.14 0.784 0.975
6 Abcc8 2576. 2609. 2292. 0.889 0.878
7 Abhd14a 591. 547. 432. 0.731 0.791
8 Abi2 4881. 4903. 4945. 1.01 1.01
9 Abi3bp 1175. 1061. 762. 0.649 0.719
10 Abl2 2170. 2078. 2131. 0.982 1.03
# ℹ 1,464 more rowsそして、pivot_longer()関数を使用する:
R
rna_time |>
mutate(time_8_vs_0 = `8` / `0`, time_8_vs_4 = `8` / `4`) |>
pivot_longer(names_to = "comparisons",
values_to = "Fold_changes",
time_8_vs_0:time_8_vs_4)
OUTPUT
# A tibble: 2,948 × 6
# Groups: gene [1,474]
gene `0` `4` `8` comparisons Fold_changes
<chr> <dbl> <dbl> <dbl> <chr> <dbl>
1 AI504432 1034. 1104. 1014 time_8_vs_0 0.981
2 AI504432 1034. 1104. 1014 time_8_vs_4 0.918
3 AW046200 155. 152. 81 time_8_vs_0 0.522
4 AW046200 155. 152. 81 time_8_vs_4 0.532
5 AW551984 238 302. 342. time_8_vs_0 1.44
6 AW551984 238 302. 342. time_8_vs_4 1.13
7 Aamp 4603. 4870 4763. time_8_vs_0 1.03
8 Aamp 4603. 4870 4763. time_8_vs_4 0.978
9 Abca12 5.29 4.25 4.14 time_8_vs_0 0.784
10 Abca12 5.29 4.25 4.14 time_8_vs_4 0.975
# ℹ 2,938 more rowsテーブルの結合
実生活の多くの場面で、データは複数のテーブルにまたがっている。 通常このようなことが起こるのは、異なる情報源から異なるタイプの情報が 収集されるからである。 通常このようなことが起こるのは、異なる情報源から異なるタイプの情報が 収集されるからである。
分析によっては、2つ以上のテーブル( )のデータを、すべてのテーブルに共通するカラム( )に基づいて1つのデータフレームにまとめることが望ましい場合がある。
dplyr\` パッケージは、指定されたカラム内のマッチに基づいて、2つの データフレームを結合するための結合関数のセットを提供する。 ここでは、 、結合について簡単に紹介する。 詳しくは、 テーブル ジョインの章を参照されたい。 データ変換チート シート 、テーブル結合に関する簡単な概要も提供している。 ここでは、 、結合について簡単に紹介する。 詳しくは、 テーブル ジョインの章を参照されたい。 データ変換チート シート 、テーブル結合に関する簡単な概要も提供している。
、元のrnaテーブルをサブセットして作成し、
、3つのカラムと10行だけを残す。
R
rna_mini <- rna |>
select(gene, sample, expression) |>
head(10)
rna_mini
OUTPUT
# A tibble: 10 × 3
gene sample expression
<chr> <chr> <dbl>
1 Asl GSM2545336 1170
2 Apod GSM2545336 36194
3 Cyp2d22 GSM2545336 4060
4 Klk6 GSM2545336 287
5 Fcrls GSM2545336 85
6 Slc2a4 GSM2545336 782
7 Exd2 GSM2545336 1619
8 Gjc2 GSM2545336 288
9 Plp1 GSM2545336 43217
10 Gnb4 GSM2545336 10712番目のテーブルannot1には、遺伝子と
gene_descriptionの2つのカラムがある。
2番目のテーブルannot1には、遺伝子と
gene_descriptionの2つのカラムがある。 download
annot1.csv
リンクをクリックしてdata/フォルダに移動するか、
以下のRコードを使って直接フォルダにダウンロードすることができる。
R
download.file(url = "https://carpentries-incubator.github.io/bioc-intro/data/annot1.csv",
destfile = "data/annot1.csv")
annot1 <- read_csv(file = "data/annot1.csv")
annot1
OUTPUT
# A tibble: 10 × 2
gene gene_description
<chr> <chr>
1 Cyp2d22 cytochrome P450, family 2, subfamily d, polypeptide 22 [Source:MGI S…
2 Klk6 kallikrein related-peptidase 6 [Source:MGI Symbol;Acc:MGI:1343166]
3 Fcrls Fc receptor-like S, scavenger receptor [Source:MGI Symbol;Acc:MGI:19…
4 Plp1 proteolipid protein (myelin) 1 [Source:MGI Symbol;Acc:MGI:97623]
5 Exd2 exonuclease 3'-5' domain containing 2 [Source:MGI Symbol;Acc:MGI:192…
6 Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88056]
7 Gnb4 guanine nucleotide binding protein (G protein), beta 4 [Source:MGI S…
8 Slc2a4 solute carrier family 2 (facilitated glucose transporter), member 4 …
9 Asl argininosuccinate lyase [Source:MGI Symbol;Acc:MGI:88084]
10 Gjc2 gap junction protein, gamma 2 [Source:MGI Symbol;Acc:MGI:2153060] ここで、dplyr パッケージの full_join()
関数を使用して、これら2つのテーブルを、すべての
変数を含む1つのテーブルに結合したいと思います。
関数は、最初のテーブルと2番目のテーブルの列
に一致する共通変数を自動的に見つける。
この場合、geneは共通の 。 このような変数をキーと呼ぶ。
キーは、
オブザベーションを異なるテーブル間でマッチさせるために使用される。
関数は、最初のテーブルと2番目のテーブルの列
に一致する共通変数を自動的に見つける。
この場合、geneは共通の 。 このような変数をキーと呼ぶ。
キーは、
オブザベーションを異なるテーブル間でマッチさせるために使用される。
R
full_join(rna_mini, annot1)
OUTPUT
Joining with `by = join_by(gene)`OUTPUT
# A tibble: 10 × 4
gene sample expression gene_description
<chr> <chr> <dbl> <chr>
1 Asl GSM2545336 1170 argininosuccinate lyase [Source:MGI Symbol;Acc…
2 Apod GSM2545336 36194 apolipoprotein D [Source:MGI Symbol;Acc:MGI:88…
3 Cyp2d22 GSM2545336 4060 cytochrome P450, family 2, subfamily d, polype…
4 Klk6 GSM2545336 287 kallikrein related-peptidase 6 [Source:MGI Sym…
5 Fcrls GSM2545336 85 Fc receptor-like S, scavenger receptor [Source…
6 Slc2a4 GSM2545336 782 solute carrier family 2 (facilitated glucose t…
7 Exd2 GSM2545336 1619 exonuclease 3'-5' domain containing 2 [Source:…
8 Gjc2 GSM2545336 288 gap junction protein, gamma 2 [Source:MGI Symb…
9 Plp1 GSM2545336 43217 proteolipid protein (myelin) 1 [Source:MGI Sym…
10 Gnb4 GSM2545336 1071 guanine nucleotide binding protein (G protein)…実生活では、遺伝子アノテーションのラベルが異なることがある。
annot2テーブルは、遺伝子名を含む 変数のラベルが異なる以外は、annot1と全く同じである。 この場合も、 [download annot2.csv](https://carpentries-incubator.github.io/bioc-intro/data/annot2.csv) 、自分でdata/`に移動するか、以下のRコードを使う。
この場合も、 download
annot2.csv
、自分でdata/\に移動するか、以下のRコードを使う。
R
download.file(url = "https://carpentries-incubator.github.io/bioc-intro/data/annot2.csv",
destfile = "data/annot2.csv")
annot2 <- read_csv(file = "data/annot2.csv")
annot2
OUTPUT
# A tibble: 10 × 2
external_gene_name description
<chr> <chr>
1 Cyp2d22 cytochrome P450, family 2, subfamily d, polypeptide 22 [S…
2 Klk6 kallikrein related-peptidase 6 [Source:MGI Symbol;Acc:MGI…
3 Fcrls Fc receptor-like S, scavenger receptor [Source:MGI Symbol…
4 Plp1 proteolipid protein (myelin) 1 [Source:MGI Symbol;Acc:MGI…
5 Exd2 exonuclease 3'-5' domain containing 2 [Source:MGI Symbol;…
6 Apod apolipoprotein D [Source:MGI Symbol;Acc:MGI:88056]
7 Gnb4 guanine nucleotide binding protein (G protein), beta 4 [S…
8 Slc2a4 solute carrier family 2 (facilitated glucose transporter)…
9 Asl argininosuccinate lyase [Source:MGI Symbol;Acc:MGI:88084]
10 Gjc2 gap junction protein, gamma 2 [Source:MGI Symbol;Acc:MGI:…どの変数名も一致しない場合、マッチングに使用する
変数を手動で設定することができる。
どの変数名も一致しない場合、マッチングに使用する
変数を手動で設定することができる。 これらの変数は、rna_mini
と annot2 テーブルを使用して以下に示すように、
by 引数を使用して設定することができる。
R
full_join(rna_mini, annot2, by = c("gene" = "external_gene_name"))
OUTPUT
# A tibble: 10 × 4
gene sample expression description
<chr> <chr> <dbl> <chr>
1 Asl GSM2545336 1170 argininosuccinate lyase [Source:MGI Symbol;Acc…
2 Apod GSM2545336 36194 apolipoprotein D [Source:MGI Symbol;Acc:MGI:88…
3 Cyp2d22 GSM2545336 4060 cytochrome P450, family 2, subfamily d, polype…
4 Klk6 GSM2545336 287 kallikrein related-peptidase 6 [Source:MGI Sym…
5 Fcrls GSM2545336 85 Fc receptor-like S, scavenger receptor [Source…
6 Slc2a4 GSM2545336 782 solute carrier family 2 (facilitated glucose t…
7 Exd2 GSM2545336 1619 exonuclease 3'-5' domain containing 2 [Source:…
8 Gjc2 GSM2545336 288 gap junction protein, gamma 2 [Source:MGI Symb…
9 Plp1 GSM2545336 43217 proteolipid protein (myelin) 1 [Source:MGI Sym…
10 Gnb4 GSM2545336 1071 guanine nucleotide binding protein (G protein)…上で見たように、最初のテーブルの変数名は、結合されたテーブルでも 。
チャレンジだ:
こちら
をクリックして annot3
テーブルをダウンロードし、そのテーブルをあなたの data/
リポジトリに置いてください。
full_join()関数を使用して、テーブルrna_miniとannot3`
を結合する。
、遺伝子_Klk6_、mt-Tf、mt-Rnr1、mt-Tv、mt-Rnr2、mt-Tl1_はどうなったのか?
full_join()関数を使用して、テーブルrna_miniとannot3`
を結合する。
、遺伝子_Klk6、mt-Tf、mt-Rnr1、mt-Tv、mt-Rnr2、_mt-Tl1_はどうなったのか?
R
annot3 <- read_csv("data/annot3.csv")
full_join(rna_mini, annot3)
OUTPUT
# A tibble: 15 × 4
gene sample expression gene_description
<chr> <chr> <dbl> <chr>
1 Asl GSM2545336 1170 argininosuccinate lyase [Source:MGI Symbol;Acc…
2 Apod GSM2545336 36194 apolipoprotein D [Source:MGI Symbol;Acc:MGI:88…
3 Cyp2d22 GSM2545336 4060 cytochrome P450, family 2, subfamily d, polype…
4 Klk6 GSM2545336 287 <NA>
5 Fcrls GSM2545336 85 Fc receptor-like S, scavenger receptor [Source…
6 Slc2a4 GSM2545336 782 solute carrier family 2 (facilitated glucose t…
7 Exd2 GSM2545336 1619 exonuclease 3'-5' domain containing 2 [Source:…
8 Gjc2 GSM2545336 288 gap junction protein, gamma 2 [Source:MGI Symb…
9 Plp1 GSM2545336 43217 proteolipid protein (myelin) 1 [Source:MGI Sym…
10 Gnb4 GSM2545336 1071 guanine nucleotide binding protein (G protein)…
11 mt-Tf <NA> NA mitochondrially encoded tRNA phenylalanine [So…
12 mt-Rnr1 <NA> NA mitochondrially encoded 12S rRNA [Source:MGI S…
13 mt-Tv <NA> NA mitochondrially encoded tRNA valine [Source:MG…
14 mt-Rnr2 <NA> NA mitochondrially encoded 16S rRNA [Source:MGI S…
15 mt-Tl1 <NA> NA mitochondrially encoded tRNA leucine 1 [Source…遺伝子_Klk6_はrna_miniにのみ存在し、遺伝子_mt-Tf_、mt-Rnr1、mt-Tv、
mt-Rnr2、_mt-Tl1_はannot3テーブルにのみ存在する。
表の 変数のそれぞれの値は、欠損として符号化されている。 表の
変数のそれぞれの値は、欠損として符号化されている。
データのエクスポート
dplyr\`を使って、 から情報を抽出したり、生データを要約したりする方法を学んだので、これらの新しいデータセットをエクスポートして、 を共同研究者と共有したり、アーカイブしたりしたいと思うかもしれない。
RにCSVファイルを読み込むために使用される read_csv()
関数と同様に、 、データフレームからCSVファイルを生成する
write_csv() 関数があります。
write_csv()を使う前に、生成されたデータセットを格納する新しいフォルダdata_outputを作業ディレクトリに作成する。 、生成されたデータセットを生データと同じディレクトリに書き込みたくない。 別々にするのは良い習慣だ。 dataフォルダーには、
、変更されていない生のデータだけを入れておく。
、削除したり変更したりしないように、そのままにしておく。
対照的に、このスクリプトはdata_output
ディレクトリの内容を生成するので、そこに含まれるファイルが削除されても、
再生成することができる。
、生成されたデータセットを生データと同じディレクトリに書き込みたくない。
別々にするのは良い習慣だ。
dataフォルダーには、 、変更されていない生のデータだけを入れておく。 、削除したり変更したりしないように、そのままにしておく。 対照的に、このスクリプトはdata_output`
ディレクトリの内容を生成するので、そこに含まれるファイルが削除されても、
再生成することができる。
write_csv()\`を使用して、以前に作成したrna_wideテーブルを保存しよう。
R
write_csv(rna_wide, file = "data_output/rna_wide.csv")
- tidyverseメタパッケージを使用したRでの表形式データ