次のステップ
Last updated on 2026-04-28 | Edit this page
Overview
Questions
-
SummarizedExperimentとは何でしょうか? - Bioconductor と何でしょうか?
Objectives
- Bioconductorプロジェクトを紹介してみましょう。
- データコンテナの概念を紹介してみましょう。
- オミックス解析で多用される
SummarizedExperimentの概要を説明する。
次のステップ
バイオインフォマティクスのデータはしばしば複雑です。 これに対処するため、 開発者は、扱う必要のあるデータのプロパティに マッチする、特別なデータコンテナ(クラスと呼ばれる)を定義する。
この側面は、パッケージ間で同じコア・データ・インフラを使用するバイオコンダクター[^バイオコンダクター]プロジェクト 。 この 、Bioconductorの成功に貢献したことは間違いない。 Bioconductor パッケージ 開発者は、 プロジェクト全体に一貫性、相互運用性、安定性を提供するために、既存のインフラストラクチャを利用することをお勧めします 。
このようなオミックス・データ・コンテナを説明するために、
SummarizedExperimentクラスを紹介する。
実験概要
下図は、SummarizedExperimentクラスの構造を表しています。

SummarizedExperimentクラスのオブジェクトには、:
**定量的オミックスデータ (発現データ)を含む1つ(または複数)のアッセイ **、マトリックス状のオブジェクトとして格納されている。 特徴(遺伝子、 転写物、タンパク質、…) は行に沿って定義され、 は列に沿って定義される。
データフレームとして格納された、サンプルの共変量を含む sample metadata スロット。 この表の行はサンプルを表す(行は発現データの 列と正確に一致する)。
データフレームとして格納される、特徴共変量を含む 特徴メタデータ スロット。 このデータフレームの行は、 式データの行と完全に一致する。
SummarizedExperiment`の調整された性質は、データ操作中に 、異なるスロットの次元が 、常に一致することを保証する(すなわち、発現データの列と サンプルメタデータの行、および発現データと 特徴メタデータの行)。 例えば、 、アッセイから1つのサンプルを除外しなければならない場合、同じ操作でサンプルメタデータから 、自動的に除外される。
メタデータ・スロットは、他の構造に影響を与えることなく、 (カラム)の共変数を追加で増やすことができる。
SummarizedExperimentの作成
SummarizedExperiment`を作成するために、 の各コンポーネント、すなわちカウントマトリックス、サンプル、遺伝子 のメタデータをcsvファイルから作成する。 これらは通常、RNA-Seqデータが (生データが処理された後)提供される方法である。
-
An expression matrix: カウント行列をロードし、
最初の列が行/遺伝子名を含むことを指定し、
data.frameをmatrixに変換する。 ダウンロードは こちら。
R
count_matrix <- read.csv("data/count_matrix.csv",
row.names = 1) %>%
as.matrix()
count_matrix[1:5, ]
OUTPUT
GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
Asl 1170 361 400 586 626 988
Apod 36194 10347 9173 10620 13021 29594
Cyp2d22 4060 1616 1603 1901 2171 3349
Klk6 287 629 641 578 448 195
Fcrls 85 233 244 237 180 38
GSM2545342 GSM2545343 GSM2545344 GSM2545345 GSM2545346 GSM2545347
Asl 836 535 586 597 938 1035
Apod 24959 13668 13230 15868 27769 34301
Cyp2d22 3122 2008 2254 2277 2985 3452
Klk6 186 1101 537 567 327 233
Fcrls 68 375 199 177 89 67
GSM2545348 GSM2545349 GSM2545350 GSM2545351 GSM2545352 GSM2545353
Asl 494 481 666 937 803 541
Apod 11258 11812 15816 29242 20415 13682
Cyp2d22 1883 2014 2417 3678 2920 2216
Klk6 742 881 828 250 798 710
Fcrls 300 233 231 81 303 285
GSM2545354 GSM2545362 GSM2545363 GSM2545380
Asl 473 748 576 1192
Apod 11088 15916 11166 38148
Cyp2d22 1821 2842 2011 4019
Klk6 894 501 598 259
Fcrls 248 179 184 68R
dim(count_matrix)
OUTPUT
[1] 1474 22- サンプルを説明する表、 こちら。
R
sample_metadata <- read.csv("data/sample_metadata.csv")
sample_metadata
OUTPUT
sample organism age sex infection strain time tissue mouse
1 GSM2545336 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 14
2 GSM2545337 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 9
3 GSM2545338 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 10
4 GSM2545339 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 15
5 GSM2545340 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 18
6 GSM2545341 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum 6
7 GSM2545342 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 5
8 GSM2545343 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum 11
9 GSM2545344 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 22
10 GSM2545345 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 13
11 GSM2545346 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum 23
12 GSM2545347 Mus musculus 8 Male InfluenzaA C57BL/6 8 Cerebellum 24
13 GSM2545348 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 8
14 GSM2545349 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum 7
15 GSM2545350 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 1
16 GSM2545351 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 16
17 GSM2545352 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 21
18 GSM2545353 Mus musculus 8 Female NonInfected C57BL/6 0 Cerebellum 4
19 GSM2545354 Mus musculus 8 Male NonInfected C57BL/6 0 Cerebellum 2
20 GSM2545362 Mus musculus 8 Female InfluenzaA C57BL/6 4 Cerebellum 20
21 GSM2545363 Mus musculus 8 Male InfluenzaA C57BL/6 4 Cerebellum 12
22 GSM2545380 Mus musculus 8 Female InfluenzaA C57BL/6 8 Cerebellum 19R
dim(sample_metadata)
OUTPUT
[1] 22 9- 遺伝子を説明する表、 こちら。
R
gene_metadata <- read.csv("data/gene_metadata.csv")
gene_metadata[1:10, 1:4]
OUTPUT
gene ENTREZID
1 Asl 109900
2 Apod 11815
3 Cyp2d22 56448
4 Klk6 19144
5 Fcrls 80891
6 Slc2a4 20528
7 Exd2 97827
8 Gjc2 118454
9 Plp1 18823
10 Gnb4 14696
product
1 argininosuccinate lyase, transcript variant X1
2 apolipoprotein D, transcript variant 3
3 cytochrome P450, family 2, subfamily d, polypeptide 22, transcript variant 2
4 kallikrein related-peptidase 6, transcript variant 2
5 Fc receptor-like S, scavenger receptor, transcript variant X1
6 solute carrier family 2 (facilitated glucose transporter), member 4
7 exonuclease 3'-5' domain containing 2
8 gap junction protein, gamma 2, transcript variant 1
9 proteolipid protein (myelin) 1, transcript variant 1
10 guanine nucleotide binding protein (G protein), beta 4, transcript variant X2
ensembl_gene_id
1 ENSMUSG00000025533
2 ENSMUSG00000022548
3 ENSMUSG00000061740
4 ENSMUSG00000050063
5 ENSMUSG00000015852
6 ENSMUSG00000018566
7 ENSMUSG00000032705
8 ENSMUSG00000043448
9 ENSMUSG00000031425
10 ENSMUSG00000027669R
dim(gene_metadata)
OUTPUT
[1] 1474 9これらのテーブルから SummarizedExperiment
を作成する:
として使用されるカウント行列。
サンプルを記述したテーブルは、サンプル メタデータスロットとして使用される。
遺伝子を記述したテーブルは、features メタデータスロットとして使用される。
これを行うには、 SummarizedExperiment
コンストラクタを使って、異なるパーツをまとめることができる:
R
## BiocManager::install("SummarizedExperiment")
library("SummarizedExperiment")
まず、 カウントマトリックスとサンプルアノテーションにおいて、サンプルの順番が同じであることを確認する。また、 カウントマトリックスと遺伝子アノテーションにおいて、遺伝子の順番が同じであることを確認する。
R
stopifnot(rownames(count_matrix) == gene_metadata$gene)
stopifnot(colnames(count_matrix) == sample_metadata$sample)
R
se <- SummarizedExperiment(assays = list(counts = count_matrix),
colData = sample_metadata,
rowData = gene_metadata)
se
OUTPUT
class: SummarizedExperiment
dim: 1474 22
metadata(0):
assays(1): counts
rownames(1474): Asl Apod ... Lmx1a Pbx1
rowData names(9): gene ENTREZID ... phenotype_description
hsapiens_homolog_associated_gene_name
colnames(22): GSM2545336 GSM2545337 ... GSM2545363 GSM2545380
colData names(9): sample organism ... tissue mouseデータの保存
以前のエピソードで行ったように、データをスプレッドシートにエクスポートするには、
、第1章
(小数点以下の区切り文字に,と.を使った場合の不整合の可能性、
変数型の定義の欠如)で説明したようないくつかの制限がある。 さらに、
スプレッドシートへのデータエクスポートは、データフレーム
や行列のような長方形のデータにのみ関係する。
データを保存するより一般的な方法は、Rに特有であり、
どのオペレーティングシステムでも動作することが保証されている
saveRDS 関数を使用することである。
このようにオブジェクトを保存すると、ディスク上にバイナリ
表現が生成されます(ここでは rds
ファイル拡張子を使用します)。 readRDS 関数を使用して R
にロードし直すことができます。
R
saveRDS(se, file = "data_output/se.rds")
rm(se)
se <- readRDS("data_output/se.rds")
head(se)
結論として、Rからデータを保存し、
Rで再度ロードする場合、saveRDSとreadRDSで保存とロードを行うのが
。 表形式のデータを、Rを使用していない誰か(
)と共有する必要がある場合は、テキストベースのスプレッドシートにエクスポートするのが、
良い選択肢である。
このデータ構造を使って、
assay関数で発現行列にアクセスすることができる:
R
head(assay(se))
OUTPUT
GSM2545336 GSM2545337 GSM2545338 GSM2545339 GSM2545340 GSM2545341
Asl 1170 361 400 586 626 988
Apod 36194 10347 9173 10620 13021 29594
Cyp2d22 4060 1616 1603 1901 2171 3349
Klk6 287 629 641 578 448 195
Fcrls 85 233 244 237 180 38
Slc2a4 782 231 248 265 313 786
GSM2545342 GSM2545343 GSM2545344 GSM2545345 GSM2545346 GSM2545347
Asl 836 535 586 597 938 1035
Apod 24959 13668 13230 15868 27769 34301
Cyp2d22 3122 2008 2254 2277 2985 3452
Klk6 186 1101 537 567 327 233
Fcrls 68 375 199 177 89 67
Slc2a4 528 249 266 357 654 693
GSM2545348 GSM2545349 GSM2545350 GSM2545351 GSM2545352 GSM2545353
Asl 494 481 666 937 803 541
Apod 11258 11812 15816 29242 20415 13682
Cyp2d22 1883 2014 2417 3678 2920 2216
Klk6 742 881 828 250 798 710
Fcrls 300 233 231 81 303 285
Slc2a4 271 304 349 715 513 320
GSM2545354 GSM2545362 GSM2545363 GSM2545380
Asl 473 748 576 1192
Apod 11088 15916 11166 38148
Cyp2d22 1821 2842 2011 4019
Klk6 894 501 598 259
Fcrls 248 179 184 68
Slc2a4 248 350 317 796R
dim(assay(se))
OUTPUT
[1] 1474 22colData`関数を使ってサンプルのメタデータにアクセスすることができる:
R
colData(se)
OUTPUT
DataFrame with 22 rows and 9 columns
sample organism age sex infection
<character> <character> <integer> <character> <character>
GSM2545336 GSM2545336 Mus musculus 8 Female InfluenzaA
GSM2545337 GSM2545337 Mus musculus 8 Female NonInfected
GSM2545338 GSM2545338 Mus musculus 8 Female NonInfected
GSM2545339 GSM2545339 Mus musculus 8 Female InfluenzaA
GSM2545340 GSM2545340 Mus musculus 8 Male InfluenzaA
... ... ... ... ... ...
GSM2545353 GSM2545353 Mus musculus 8 Female NonInfected
GSM2545354 GSM2545354 Mus musculus 8 Male NonInfected
GSM2545362 GSM2545362 Mus musculus 8 Female InfluenzaA
GSM2545363 GSM2545363 Mus musculus 8 Male InfluenzaA
GSM2545380 GSM2545380 Mus musculus 8 Female InfluenzaA
strain time tissue mouse
<character> <integer> <character> <integer>
GSM2545336 C57BL/6 8 Cerebellum 14
GSM2545337 C57BL/6 0 Cerebellum 9
GSM2545338 C57BL/6 0 Cerebellum 10
GSM2545339 C57BL/6 4 Cerebellum 15
GSM2545340 C57BL/6 4 Cerebellum 18
... ... ... ... ...
GSM2545353 C57BL/6 0 Cerebellum 4
GSM2545354 C57BL/6 0 Cerebellum 2
GSM2545362 C57BL/6 4 Cerebellum 20
GSM2545363 C57BL/6 4 Cerebellum 12
GSM2545380 C57BL/6 8 Cerebellum 19R
dim(colData(se))
OUTPUT
[1] 22 9また、rowData関数を使ってフィーチャーのメタデータにアクセスすることもできる:
R
head(rowData(se))
OUTPUT
DataFrame with 6 rows and 9 columns
gene ENTREZID product ensembl_gene_id
<character> <integer> <character> <character>
Asl Asl 109900 argininosuccinate ly.. ENSMUSG00000025533
Apod Apod 11815 apolipoprotein D, tr.. ENSMUSG00000022548
Cyp2d22 Cyp2d22 56448 cytochrome P450, fam.. ENSMUSG00000061740
Klk6 Klk6 19144 kallikrein related-p.. ENSMUSG00000050063
Fcrls Fcrls 80891 Fc receptor-like S, .. ENSMUSG00000015852
Slc2a4 Slc2a4 20528 solute carrier famil.. ENSMUSG00000018566
external_synonym chromosome_name gene_biotype phenotype_description
<character> <character> <character> <character>
Asl 2510006M18Rik 5 protein_coding abnormal circulating..
Apod NA 16 protein_coding abnormal lipid homeo..
Cyp2d22 2D22 15 protein_coding abnormal skin morpho..
Klk6 Bssp 7 protein_coding abnormal cytokine le..
Fcrls 2810439C17Rik 3 protein_coding decreased CD8-positi..
Slc2a4 Glut-4 11 protein_coding abnormal circulating..
hsapiens_homolog_associated_gene_name
<character>
Asl ASL
Apod APOD
Cyp2d22 CYP2D6
Klk6 KLK6
Fcrls FCRL2
Slc2a4 SLC2A4R
dim(rowData(se))
OUTPUT
[1] 1474 9SummarizedExperimentをサブセットする
SummarizedExperiment は、データフレームと同じように、 数値または論理の文字でサブセットできる。
以下では、 、3つの最初のサンプルの5つの最初の特徴のみを含む、SummarizedExperimentクラスの新しいインスタンスを作成します。
R
se1 <- se[1:5, 1:3]
se1
OUTPUT
class: SummarizedExperiment
dim: 5 3
metadata(0):
assays(1): counts
rownames(5): Asl Apod Cyp2d22 Klk6 Fcrls
rowData names(9): gene ENTREZID ... phenotype_description
hsapiens_homolog_associated_gene_name
colnames(3): GSM2545336 GSM2545337 GSM2545338
colData names(9): sample organism ... tissue mouseR
colData(se1)
OUTPUT
DataFrame with 3 rows and 9 columns
sample organism age sex infection
<character> <character> <integer> <character> <character>
GSM2545336 GSM2545336 Mus musculus 8 Female InfluenzaA
GSM2545337 GSM2545337 Mus musculus 8 Female NonInfected
GSM2545338 GSM2545338 Mus musculus 8 Female NonInfected
strain time tissue mouse
<character> <integer> <character> <integer>
GSM2545336 C57BL/6 8 Cerebellum 14
GSM2545337 C57BL/6 0 Cerebellum 9
GSM2545338 C57BL/6 0 Cerebellum 10R
rowData(se1)
OUTPUT
DataFrame with 5 rows and 9 columns
gene ENTREZID product ensembl_gene_id
<character> <integer> <character> <character>
Asl Asl 109900 argininosuccinate ly.. ENSMUSG00000025533
Apod Apod 11815 apolipoprotein D, tr.. ENSMUSG00000022548
Cyp2d22 Cyp2d22 56448 cytochrome P450, fam.. ENSMUSG00000061740
Klk6 Klk6 19144 kallikrein related-p.. ENSMUSG00000050063
Fcrls Fcrls 80891 Fc receptor-like S, .. ENSMUSG00000015852
external_synonym chromosome_name gene_biotype phenotype_description
<character> <character> <character> <character>
Asl 2510006M18Rik 5 protein_coding abnormal circulating..
Apod NA 16 protein_coding abnormal lipid homeo..
Cyp2d22 2D22 15 protein_coding abnormal skin morpho..
Klk6 Bssp 7 protein_coding abnormal cytokine le..
Fcrls 2810439C17Rik 3 protein_coding decreased CD8-positi..
hsapiens_homolog_associated_gene_name
<character>
Asl ASL
Apod APOD
Cyp2d22 CYP2D6
Klk6 KLK6
Fcrls FCRL2また、colData() 関数を使用して、
サンプルメタデータから何かをサブセットしたり、rowData()
関数を使用して、
フィーチャーメタデータから何かをサブセットすることもできます。
例えば、ここではmiRNAと に感染していないサンプルだけを残している:
R
se1 <- se[rowData(se)$gene_biotype == "miRNA",
colData(se)$infection == "NonInfected"]
se1
OUTPUT
class: SummarizedExperiment
dim: 7 7
metadata(0):
assays(1): counts
rownames(7): Mir1901 Mir378a ... Mir128-1 Mir7682
rowData names(9): gene ENTREZID ... phenotype_description
hsapiens_homolog_associated_gene_name
colnames(7): GSM2545337 GSM2545338 ... GSM2545353 GSM2545354
colData names(9): sample organism ... tissue mouseR
assay(se1)
OUTPUT
GSM2545337 GSM2545338 GSM2545343 GSM2545348 GSM2545349 GSM2545353
Mir1901 45 44 74 55 68 33
Mir378a 11 7 9 4 12 4
Mir133b 4 6 5 4 6 7
Mir30c-2 10 6 16 12 8 17
Mir149 1 2 0 0 0 0
Mir128-1 4 1 2 2 1 2
Mir7682 2 0 4 1 3 5
GSM2545354
Mir1901 60
Mir378a 8
Mir133b 3
Mir30c-2 15
Mir149 2
Mir128-1 1
Mir7682 5R
colData(se1)
OUTPUT
DataFrame with 7 rows and 9 columns
sample organism age sex infection
<character> <character> <integer> <character> <character>
GSM2545337 GSM2545337 Mus musculus 8 Female NonInfected
GSM2545338 GSM2545338 Mus musculus 8 Female NonInfected
GSM2545343 GSM2545343 Mus musculus 8 Male NonInfected
GSM2545348 GSM2545348 Mus musculus 8 Female NonInfected
GSM2545349 GSM2545349 Mus musculus 8 Male NonInfected
GSM2545353 GSM2545353 Mus musculus 8 Female NonInfected
GSM2545354 GSM2545354 Mus musculus 8 Male NonInfected
strain time tissue mouse
<character> <integer> <character> <integer>
GSM2545337 C57BL/6 0 Cerebellum 9
GSM2545338 C57BL/6 0 Cerebellum 10
GSM2545343 C57BL/6 0 Cerebellum 11
GSM2545348 C57BL/6 0 Cerebellum 8
GSM2545349 C57BL/6 0 Cerebellum 7
GSM2545353 C57BL/6 0 Cerebellum 4
GSM2545354 C57BL/6 0 Cerebellum 2R
rowData(se1)
OUTPUT
DataFrame with 7 rows and 9 columns
gene ENTREZID product ensembl_gene_id
<character> <integer> <character> <character>
Mir1901 Mir1901 100316686 microRNA 1901 ENSMUSG00000084565
Mir378a Mir378a 723889 microRNA 378a ENSMUSG00000105200
Mir133b Mir133b 723817 microRNA 133b ENSMUSG00000065480
Mir30c-2 Mir30c-2 723964 microRNA 30c-2 ENSMUSG00000065567
Mir149 Mir149 387167 microRNA 149 ENSMUSG00000065470
Mir128-1 Mir128-1 387147 microRNA 128-1 ENSMUSG00000065520
Mir7682 Mir7682 102466847 microRNA 7682 ENSMUSG00000106406
external_synonym chromosome_name gene_biotype phenotype_description
<character> <character> <character> <character>
Mir1901 Mirn1901 18 miRNA NA
Mir378a Mirn378 18 miRNA abnormal mitochondri..
Mir133b mir 133b 1 miRNA no abnormal phenotyp..
Mir30c-2 mir 30c-2 1 miRNA NA
Mir149 Mirn149 1 miRNA increased circulatin..
Mir128-1 Mirn128 1 miRNA no abnormal phenotyp..
Mir7682 mmu-mir-7682 1 miRNA NA
hsapiens_homolog_associated_gene_name
<character>
Mir1901 NA
Mir378a MIR378A
Mir133b MIR133B
Mir30c-2 MIR30C2
Mir149 NA
Mir128-1 MIR128-1
Mir7682 NAチャレンジ
時刻0と時刻8のサンプル 、最初の3遺伝子の遺伝子発現レベルを抽出する。
R
assay(se)[1:3, colData(se)$time != 4]
OUTPUT
GSM2545336 GSM2545337 GSM2545338 GSM2545341 GSM2545342 GSM2545343
Asl 1170 361 400 988 836 535
Apod 36194 10347 9173 29594 24959 13668
Cyp2d22 4060 1616 1603 3349 3122 2008
GSM2545346 GSM2545347 GSM2545348 GSM2545349 GSM2545351 GSM2545353
Asl 938 1035 494 481 937 541
Apod 27769 34301 11258 11812 29242 13682
Cyp2d22 2985 3452 1883 2014 3678 2216
GSM2545354 GSM2545380
Asl 473 1192
Apod 11088 38148
Cyp2d22 1821 4019R
# Equivalent to
assay(se)[1:3, colData(se)$time == 0 | colData(se)$time == 8]
OUTPUT
GSM2545336 GSM2545337 GSM2545338 GSM2545341 GSM2545342 GSM2545343
Asl 1170 361 400 988 836 535
Apod 36194 10347 9173 29594 24959 13668
Cyp2d22 4060 1616 1603 3349 3122 2008
GSM2545346 GSM2545347 GSM2545348 GSM2545349 GSM2545351 GSM2545353
Asl 938 1035 494 481 937 541
Apod 27769 34301 11258 11812 29242 13682
Cyp2d22 2985 3452 1883 2014 3678 2216
GSM2545354 GSM2545380
Asl 473 1192
Apod 11088 38148
Cyp2d22 1821 4019チャレンジ
長いrnaテーブルを使用して同じ値が得られることを確認する。
R
rna |>
filter(gene %in% c("Asl", "Apod", "Cyd2d22"))|>
filter(time != 4) |> select(expression)
OUTPUT
# A tibble: 28 × 1
expression
<dbl>
1 1170
2 36194
3 361
4 10347
5 400
6 9173
7 988
8 29594
9 836
10 24959
# ℹ 18 more rows長いテーブルとSummarizedExperimentは同じ
情報を含むが、単に構造が異なるだけである。 各アプローチにはそれぞれ
独自の利点がある。前者は tidyverse パッケージに適しており、
一方、後者は多くのバイオインフォマティクスと
統計処理ステップに適した構造である。 例えば、
DESeq2パッケージを使用した典型的なRNA-Seq分析である。
メタデータに変数を追加する
メタデータに情報を追加することもできる。 サンプルが採取されたセンターを追加したいとします…
R
colData(se)$center <- rep("University of Illinois", nrow(colData(se)))
colData(se)
OUTPUT
DataFrame with 22 rows and 10 columns
sample organism age sex infection
<character> <character> <integer> <character> <character>
GSM2545336 GSM2545336 Mus musculus 8 Female InfluenzaA
GSM2545337 GSM2545337 Mus musculus 8 Female NonInfected
GSM2545338 GSM2545338 Mus musculus 8 Female NonInfected
GSM2545339 GSM2545339 Mus musculus 8 Female InfluenzaA
GSM2545340 GSM2545340 Mus musculus 8 Male InfluenzaA
... ... ... ... ... ...
GSM2545353 GSM2545353 Mus musculus 8 Female NonInfected
GSM2545354 GSM2545354 Mus musculus 8 Male NonInfected
GSM2545362 GSM2545362 Mus musculus 8 Female InfluenzaA
GSM2545363 GSM2545363 Mus musculus 8 Male InfluenzaA
GSM2545380 GSM2545380 Mus musculus 8 Female InfluenzaA
strain time tissue mouse center
<character> <integer> <character> <integer> <character>
GSM2545336 C57BL/6 8 Cerebellum 14 University of Illinois
GSM2545337 C57BL/6 0 Cerebellum 9 University of Illinois
GSM2545338 C57BL/6 0 Cerebellum 10 University of Illinois
GSM2545339 C57BL/6 4 Cerebellum 15 University of Illinois
GSM2545340 C57BL/6 4 Cerebellum 18 University of Illinois
... ... ... ... ... ...
GSM2545353 C57BL/6 0 Cerebellum 4 University of Illinois
GSM2545354 C57BL/6 0 Cerebellum 2 University of Illinois
GSM2545362 C57BL/6 4 Cerebellum 20 University of Illinois
GSM2545363 C57BL/6 4 Cerebellum 12 University of Illinois
GSM2545380 C57BL/6 8 Cerebellum 19 University of Illinoisこれは、メタデータ・スロットが、 、他の構造に影響を与えることなく、無限に成長できることを示している!
tidySummarizedExperiment
SummarizedExperiment オブジェクトを操作するために
tidyverse コマンドを使うことはできるのだろうか?
tidySummarizedExperiment パッケージを使えば可能です。
SummarizedExperimentオブジェクトがどのようなものか思い出してください:
R
シー
ERROR
Error: object 'シー' not foundtidySummarizedExperiment`をロードし、seオブジェクト 。
R
#BiocManager::install("tidySummarizedExperiment")
library("tidySummarizedExperiment")
se
WARNING
Warning: `when()` was deprecated in purrr 1.0.0.
ℹ Please use `if` instead.
ℹ The deprecated feature was likely used in the tidySummarizedExperiment
package.
Please report the issue at
<https://github.com/stemangiola/tidySummarizedExperiment/issues>.
This warning is displayed once per session.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.OUTPUT
# A SummarizedExperiment-tibble abstraction: 32,428 × 22
# Features=1474 | Samples=22 | Assays=counts
.feature .sample counts sample organism age sex infection strain time
<chr> <chr> <int> <chr> <chr> <int> <chr> <chr> <chr> <int>
1 Asl GSM2545336 1170 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
2 Apod GSM2545336 36194 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
3 Cyp2d22 GSM2545336 4060 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
4 Klk6 GSM2545336 287 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
5 Fcrls GSM2545336 85 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
6 Slc2a4 GSM2545336 782 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
7 Exd2 GSM2545336 1619 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
8 Gjc2 GSM2545336 288 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
9 Plp1 GSM2545336 43217 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
10 Gnb4 GSM2545336 1071 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
# ℹ 40 more rows
# ℹ 12 more variables: tissue <chr>, mouse <int>, center <chr>, gene <chr>,
# ENTREZID <int>, product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>これはまだSummarizedExperimentオブジェクトなので、効率的な
構造を維持しているが、これでティブルとして見ることができる。
の最初の行に注目してほしい。出力にはこう書いてあるが、これは
SummarizedExperiment-tibble の抽象化である。
また、出力の2行目には、
のトランスクリプトとサンプルの数を見ることができる。
標準のSummarizedExperimentビューに戻したい場合は、
。
R
options("restore_SummarizedExperiment_show" = TRUE)
se
OUTPUT
class: SummarizedExperiment
dim: 1474 22
metadata(0):
assays(1): counts
rownames(1474): Asl Apod ... Lmx1a Pbx1
rowData names(9): gene ENTREZID ... phenotype_description
hsapiens_homolog_associated_gene_name
colnames(22): GSM2545336 GSM2545337 ... GSM2545363 GSM2545380
colData names(10): sample organism ... mouse centerしかし、ここではティブル・ビューを使う。
R
options("restore_SummarizedExperiment_show" = FALSE)
se
OUTPUT
# A SummarizedExperiment-tibble abstraction: 32,428 × 22
# Features=1474 | Samples=22 | Assays=counts
.feature .sample counts sample organism age sex infection strain time
<chr> <chr> <int> <chr> <chr> <int> <chr> <chr> <chr> <int>
1 Asl GSM2545336 1170 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
2 Apod GSM2545336 36194 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
3 Cyp2d22 GSM2545336 4060 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
4 Klk6 GSM2545336 287 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
5 Fcrls GSM2545336 85 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
6 Slc2a4 GSM2545336 782 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
7 Exd2 GSM2545336 1619 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
8 Gjc2 GSM2545336 288 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
9 Plp1 GSM2545336 43217 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
10 Gnb4 GSM2545336 1071 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
# ℹ 40 more rows
# ℹ 12 more variables: tissue <chr>, mouse <int>, center <chr>, gene <chr>,
# ENTREZID <int>, product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>SummarizedExperiment
オブジェクトと対話するために、tidyverse
コマンドを使用できるようになりました。
filter`を使用すると、条件を使って行をフィルタリングすることができる。例えば、 、あるサンプルのすべての行を表示することができる。
R
se %>% filter(.sample == "GSM2545336")
OUTPUT
# A SummarizedExperiment-tibble abstraction: 1,474 × 22
# Features=1474 | Samples=1 | Assays=counts
.feature .sample counts sample organism age sex infection strain time
<chr> <chr> <int> <chr> <chr> <int> <chr> <chr> <chr> <int>
1 Asl GSM2545336 1170 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
2 Apod GSM2545336 36194 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
3 Cyp2d22 GSM2545336 4060 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
4 Klk6 GSM2545336 287 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
5 Fcrls GSM2545336 85 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
6 Slc2a4 GSM2545336 782 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
7 Exd2 GSM2545336 1619 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
8 Gjc2 GSM2545336 288 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
9 Plp1 GSM2545336 43217 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
10 Gnb4 GSM2545336 1071 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
# ℹ 40 more rows
# ℹ 12 more variables: tissue <chr>, mouse <int>, center <chr>, gene <chr>,
# ENTREZID <int>, product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>select`を使って表示したいカラムを指定することができる。
R
se %>% select(.sample)
OUTPUT
tidySummarizedExperiment says: Key columns are missing. A data frame is returned for independent data analysis.OUTPUT
# A tibble: 32,428 × 1
.sample
<chr>
1 GSM2545336
2 GSM2545336
3 GSM2545336
4 GSM2545336
5 GSM2545336
6 GSM2545336
7 GSM2545336
8 GSM2545336
9 GSM2545336
10 GSM2545336
# ℹ 32,418 more rowsmutate`を使ってメタデータ情報を追加することができる。
R
se %>% mutate(center = "ハイデルベルク大学")
OUTPUT
# A SummarizedExperiment-tibble abstraction: 32,428 × 22
# Features=1474 | Samples=22 | Assays=counts
.feature .sample counts sample organism age sex infection strain time
<chr> <chr> <int> <chr> <chr> <int> <chr> <chr> <chr> <int>
1 Asl GSM2545336 1170 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
2 Apod GSM2545336 36194 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
3 Cyp2d22 GSM2545336 4060 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
4 Klk6 GSM2545336 287 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
5 Fcrls GSM2545336 85 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
6 Slc2a4 GSM2545336 782 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
7 Exd2 GSM2545336 1619 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
8 Gjc2 GSM2545336 288 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
9 Plp1 GSM2545336 43217 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
10 Gnb4 GSM2545336 1071 GSM25… Mus mus… 8 Fema… Influenz… C57BL… 8
# ℹ 40 more rows
# ℹ 12 more variables: tissue <chr>, mouse <int>, center <chr>, gene <chr>,
# ENTREZID <int>, product <chr>, ensembl_gene_id <chr>,
# external_synonym <chr>, chromosome_name <chr>, gene_biotype <chr>,
# phenotype_description <chr>, hsapiens_homolog_associated_gene_name <chr>tidyverseパイプ %>%
を使ってコマンドを組み合わせることもできます。
の例では、group_by と summarise
を組み合わせて、各サンプルの カウントの合計を得ることができる。
R
se %>%
group_by(.sample) %>%
summarise(total_counts=sum(counts))
OUTPUT
tidySummarizedExperiment says: A data frame is returned for independent data analysis.OUTPUT
# A tibble: 22 × 2
.sample total_counts
<chr> <int>
1 GSM2545336 3039671
2 GSM2545337 2602360
3 GSM2545338 2458618
4 GSM2545339 2500082
5 GSM2545340 2479024
6 GSM2545341 2413723
7 GSM2545342 2349728
8 GSM2545343 3105652
9 GSM2545344 2524137
10 GSM2545345 2506038
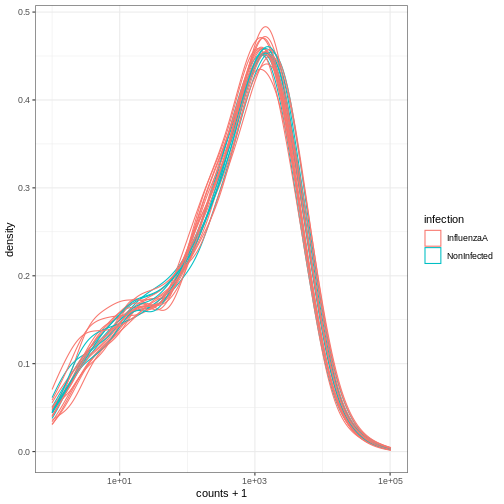
# ℹ 12 more rows整頓されたSummarizedExperimentオブジェクトを、プロット用の通常のtibble として扱うことができる。
ここでは、サンプルごとのカウント数分布をプロットしている。
R
se %>%
ggplot(aes(counts + 1, group=.sample, color=infection))+
geom_density() +
scale_x_log10() +
theme_bw()
tidySummarizedExperimentの詳細については、パッケージ ウェブサイト こちらを参照してください。
**テイクホーム・メッセージ
SummarizedExperiment`は、オミックスデータを効率的に保存し、 。
これらは多くのBioconductorパッケージで使用されている。
RNAシーケンス解析に焦点を当てた次のトレーニング、 、Bioconductor
DESeq2パッケージを使って、 差分発現解析を行う方法を学ぶ。
DESeq2パッケージの全解析はSummarizedExperiment`
で処理される。
- Bioconductorは、ハイスループットな生物学データの理解( )のためのサポートとパッケージを提供するプロジェクトである。
- SummarizedExperiment`は、ハイスループットのオミックスデータを保存し、 管理するのに便利なオブジェクトの一種である。