データの可視化
Last updated on 2026-04-28 | Edit this page
Overview
Questions
- Rによる可視化
Objectives
- ggplotを使用して散布図、箱ひげ図、折れ線グラフなどを作成できる
- プロットの共通設定を統一的に設定できる
- ファセット処理の概念を理解し、ggplotで適切に適用できる
- 既存のggplotプロットの外観要素(軸ラベルや色設定など)を変更可能である
- データフレーム内のデータから、複雑でカスタマイズ性の高いプロットを構築できる
このエピソードは、Data Carpentriesの_Data Analysis and Visualisation in R for Ecologists_レッスンに基づいています。
データの可視化
まず必要なパッケージを読み込みます。ggplot2はtidyverseパッケージに含まれています。
R
library("tidyverse")
まだワークスペースに読み込まれていない場合は、前回のレッスンで保存したデータを読み込みます。
R
rna <- read.csv("data/rnaseq.csv")
データ可視化チートシートでは、ggplot2の基本機能から高度な使い方までを網羅しており、参考資料としてだけでなく、パッケージで利用可能な多様なデータ表現方法を把握するのにも役立ちます。Thomas
Lin Pedersen氏による以下の動画チュートリアル(パート1およびパート2)も非常に参考になります。
ggplot2を使ったプロット作成
ggplot2はデータフレーム内のデータから複雑なプロットを簡単に作成できる描画パッケージです。変数の指定方法、表示方法、一般的な視覚的特性など、よりプログラム的なインターフェースを提供します。ggplot2を支える理論的基盤はグラフィックスの文法(@Wilkinson:2005)です。このアプローチを採用すれば、基礎データが変更されたり、棒グラフから散布図に変更したりする場合でも、最小限の修正で済みます。これにより、調整や微調整を最小限に抑えつつ、出版物に使用できる品質のプロットを作成できます。
ggplot2に関する書籍(@ggplot2book)も出版されていますが、内容は古めです。現在第3版が準備中で、無料でオンライン公開される予定です。ggplot2の公式ウェブサイト(https://ggplot2.tidyverse.org)には充実したドキュメントが用意されています。
ggplot2の関数は、データが「長形式」(各次元ごとに1列、各観測値ごとに1行)になっている場合に最適に動作します。適切に構造化されたデータを用意しておけば、ggplot2で図を作成する際に大幅な時間節約になります。
ggplotのグラフィックスは、新しい要素を段階的に追加することで構築されます。このようにレイヤーを追加していく方法により、プロットの表現に非常に柔軟性とカスタマイズ性が生まれます。
グラフィックスの文法の背後にある考え方は、あらゆるグラフを以下の3つの基本要素から構築できるということです:(1) データセット、(2) 座標系、(3) ジオメトリ(データポイントを表現する視覚的マーク)1
ggplotを構築するには、以下の基本的なテンプレートを使用します。これはさまざまな種類のプロットに適用できる汎用的な形式です:
ggplot(data = <DATA>, mapping = aes(<MAPPINGS>)) + <GEOM_FUNCTION>()-
ggplot()関数を使用し、data引数で特定のデータフレームにプロットを結合します
R
ggplot(data = rna)
-
マッピングを定義します(
aes関数を使用して、プロットする変数を選択し、グラフ内での表示方法を指定します。例えばx/y座標、サイズ、形状、色などの特性として表示します)
R
ggplot(data = rna, mapping = aes(x = expression))
-
’ジオメトリ’を追加します - プロット内のデータの幾何学的表現(点、線、棒など)。
ggplot2には多くの異なるジオメトリが用意されており、今日は特に一般的な以下のものを使用します:* `geom_point()` 散布図、ドットプロットなどに使用 * `geom_histogram()` ヒストグラム作成に使用 * `geom_boxplot()` ボックスプロット作成に使用 * `geom_line()` トレンドライン、時系列データなどに使用
プロットにジオメトリを追加するには+演算子を使用します。まずはgeom_histogram()を使ってみましょう:
R
ggplot(data = rna, mapping = aes(x = expression)) +
geom_histogram()
OUTPUT
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.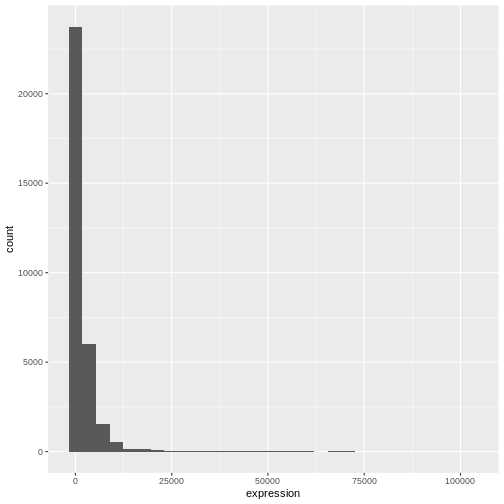
ggplot2パッケージの+演算子は特に便利で、既存のggplotオブジェクトを変更できます。つまり、プロットテンプレートを簡単に設定し、さまざまな種類のプロットを便利に探索できるため、上記のプロットは以下のようなコードでも生成可能です:
R
# プロットを変数に割り当てる
rna_plot <- ggplot(data = rna,
mapping = aes(x = expression))
# プロットを描画する
rna_plot + geom_histogram()
課題
ヒストグラムを描画する際に表示される自動メッセージにお気づきでしょうか?
OUTPUT
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.geom_histogram() の bins または
binwidth
引数を変更することで、ビンの数や幅を調整できます。
R
# ビン幅の変更
ggplot(rna, aes(x = expression)) +
geom_histogram(bins = 15)
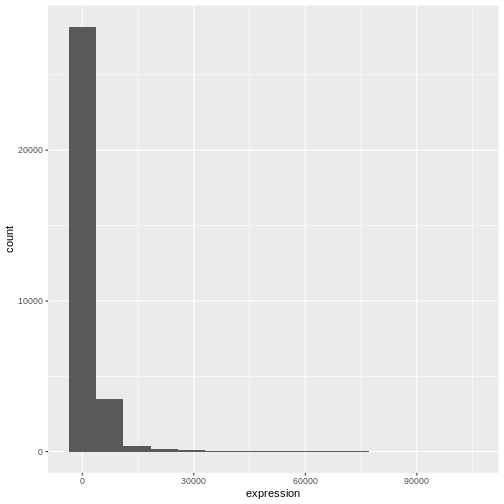
R
# ビン幅の変更
ggplot(rna, aes(x = expression)) +
geom_histogram(binwidth = 2000)
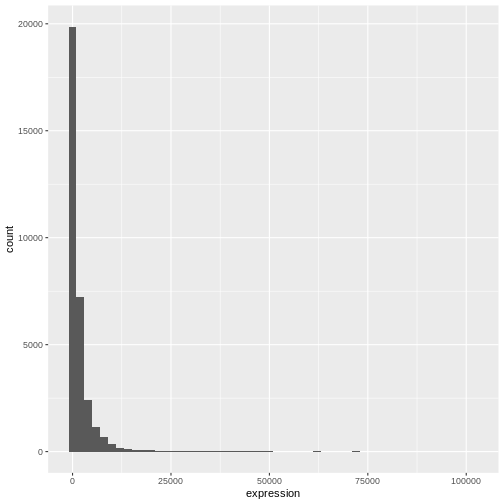
ここでデータが右に歪んでいることが確認できます。より対称的な分布を得るために、
対数変換(log2変換)を適用することができます。なお、式値が0の場合に-Inf値が
返されるのを防ぐため、ここでは小さな定数値(+1)を追加しています。
R
rna <- rna |>
mutate(expression_log = log2(expression + 1))
次に、対数変換した発現量のヒストグラムを作成すると、確かに正規分布に近い分布が得られます。
R
ggplot(rna, aes(x = expression_log)) + geom_histogram()
OUTPUT
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.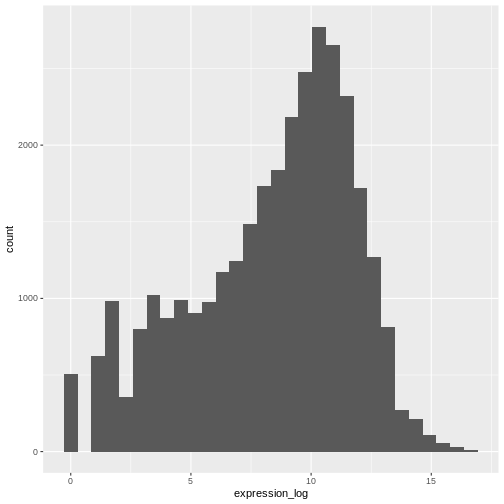
これ以降の分析では、対数変換した発現量の値を用いて作業を進めていきます。
課題
この変換を視覚的に理解する別の方法として、観測値のスケールを考慮する方法があります。例えば、軸のスケールを調整することで、プロット空間内での観測値の分布をより適切に調整できる場合があります。軸のスケール変更は、他のコンポーネントを追加・修正する場合と同様の手順で行います(つまり、段階的にコマンドを追加していく方法です)。以下の変更を試してみてください:
- 変換前の式を対数スケール(log10)で表示します。
scale_x_log10()関数を参照してください。以前のグラフと比較してください。なぜ今回は警告メッセージが表示されるのでしょうか?
R
ggplot(data = rna, mapping = aes(x = expression))+
geom_histogram() +
scale_x_log10()
WARNING
Warning in scale_x_log10(): log-10 transformation introduced infinite values.OUTPUT
`stat_bin()` using `bins = 30`. Pick better value with `binwidth`.WARNING
Warning: Removed 507 rows containing non-finite outside the scale range
(`stat_bin()`).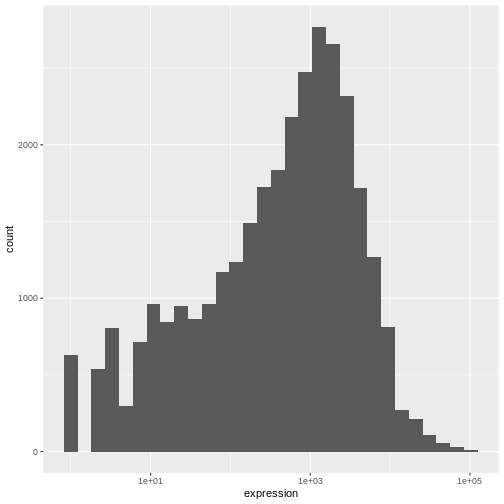
注意事項
-
ggplot()関数内で指定した設定は、後から追加するすべてのジオメトリレイヤーから参照されます(つまり、これらはグローバルなプロット設定となります)。これにはaes()で設定したx軸・y軸のマッピングも含まれます。 - 特定のジオメトリレイヤーに対しては、
ggplot()関数で定義したグローバル設定とは独立してマッピングを指定することも可能です。 - 新しいレイヤーを追加する際に使用する
+記号は、必ず直前のレイヤーを含む行の末尾に配置する必要があります。もし新しいレイヤーを含む行の先頭に+記号を置いた場合、ggplot2は新しいレイヤーを追加せず、エラーメッセージを返します。
R
# これはレイヤーを追加する正しい構文です
rna_plot +
geom_histogram()
# これは新しいレイヤーを追加せず、エラーメッセージを返します
rna_plot
+ geom_histogram()
プロットを段階的に構築する方法
ここでは、2つの連続変数を用いて散布図を作成し、geom_point()関数で表現します。このグラフは、時間8時点と時間0時点、および時間4時点と時間0時点における発現量の対数2倍変化を表します。まず、遺伝子ごと・時間ごとに対数変換した発現量の平均値を計算し、次に時間8時点と時間0時点、時間4時点と時間0時点の平均値の対数差を計算して対数2倍変化を求めます。なお、後で遺伝子を分類するために使用する遺伝子のバイオタイプ情報もここで含めます。これらの変化量は新しいデータフレームrna_fcに保存します。
R
rna_fc <- rna |> select(gene, time,
gene_biotype, expression_log) |>
group_by(gene, time, gene_biotype) |>
summarize(mean_exp = mean(expression_log)) |>
pivot_wider(names_from = time,
values_from = mean_exp) |>
mutate(time_8_vs_0 = `8` - `0`, time_4_vs_0 = `4` - `0`)
OUTPUT
`summarise()` has grouped output by 'gene', 'time'. You can override using the
`.groups` argument.作成したデータフレームrna_fcを用いてggplotを構築します。ggplot2でプロットを作成する際は通常、反復的なプロセスで行います。まず使用するデータセットを定義し、軸を設定し、使用するジオメトリを選択します:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point()
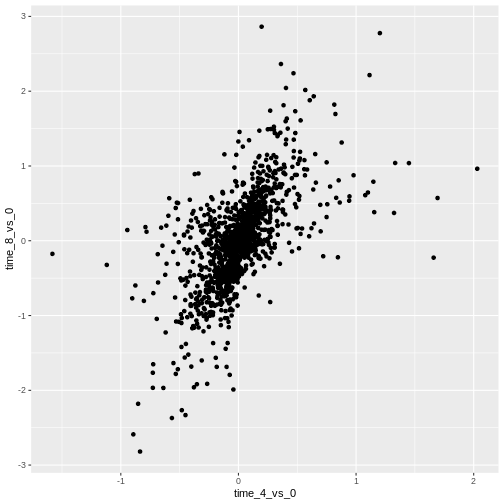
次に、このプロットをさらに修正してより多くの情報を抽出します。例えば、重なりを避けるための透明度(alpha)を追加できます:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3)
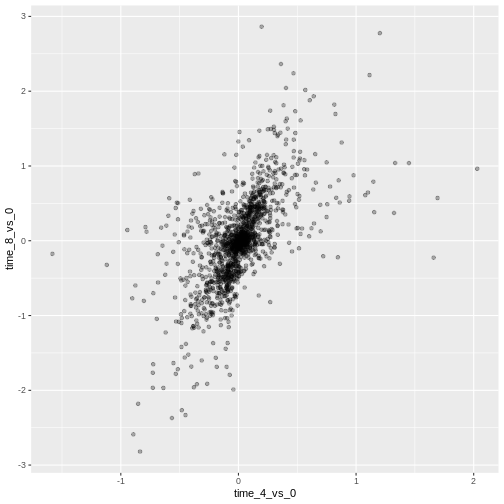
すべての点に色を付けることも可能です:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3, color = "blue")
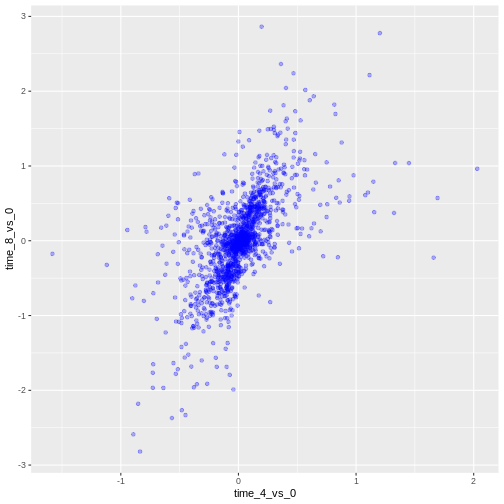
あるいは、プロット内の各遺伝子を異なる色で表示したい場合は、color引数にベクトルを指定します。ggplot2はこのベクトルの値に応じて異なる色を自動的に割り当てます。以下はgene_biotypeで色分けする例です:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_point(alpha = 0.3, aes(color = gene_biotype))
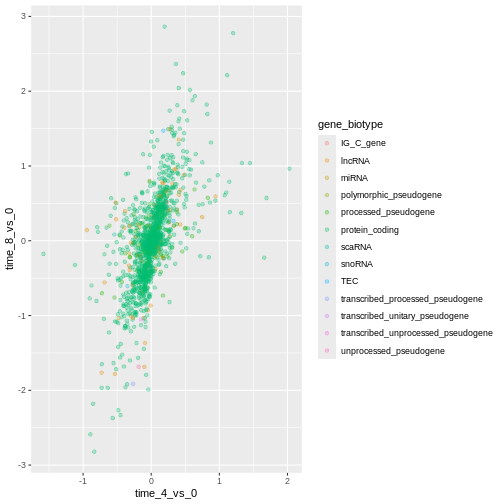
ggplot()関数内で直接マッピング時に色を指定することもできます。この設定はすべてのジオメトリレイヤーから参照され、aes()で設定したx軸・y軸のマッピングに基づいて決定されます。
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_point(alpha = 0.3)
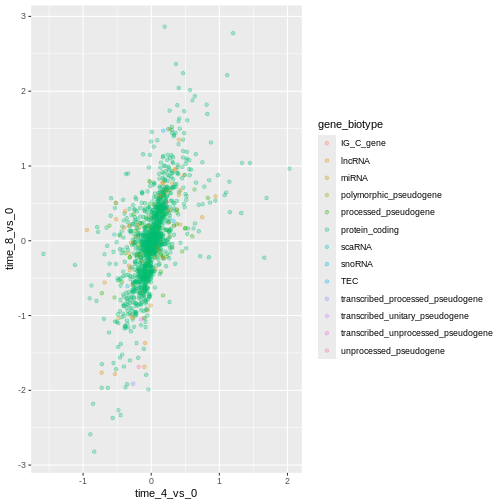
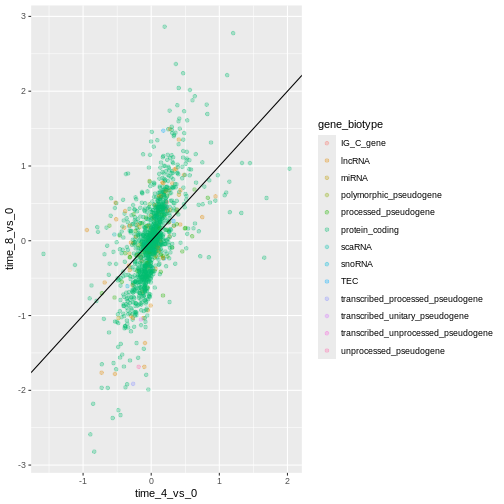
最後に、geom_abline()関数を使用して対角線を追加することも可能です:
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_point(alpha = 0.3) +
geom_abline(intercept = 0)
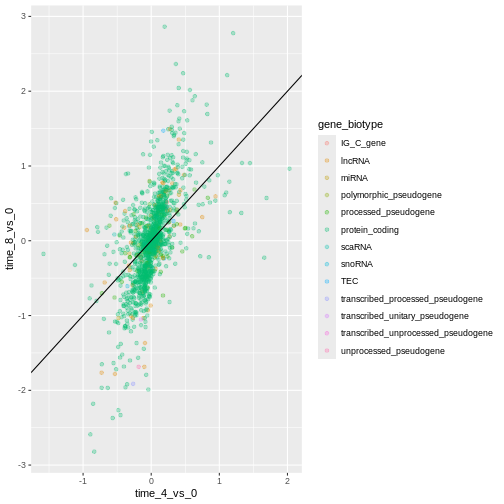
注目すべきは、ジオメトリレイヤーをgeom_pointからgeom_jitterに変更しても、色は依然としてgene_biotypeによって決定される点です。
R
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0,
color = gene_biotype)) +
geom_jitter(alpha = 0.3) +
geom_abline(intercept = 0)
課題
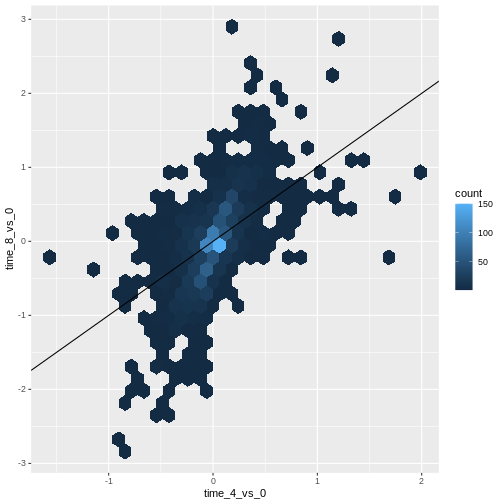
散布図は小規模なデータセットの探索的分析において有用なツールです。しかし、rna_fc
データセットのように観測値数が多い場合、点の重なり(オーバープロット)が散布図の限界となることがあります。このような状況に対処するための有効な手法の一つが、観測値を六角形グリッドに分割するヘキサビン法です。
ggplot2でヘキサビン法を使用するには、まず CRAN リポジトリから R パッケージhexbinをインストールし、ロードする必要があります。次に
geom_hex()関数を使用することで、ヘキサビンプロットを作成できます。ヘキサビンプロットと従来の散布図を比較した場合、それぞれの長所と短所は何でしょうか?上記の散布図と作成したヘキサビンプロットを詳細に比較・検討してください。
R
install.packages("hexbin")
R
library("hexbin")
ggplot(data = rna_fc, mapping = aes(x = time_4_vs_0, y = time_8_vs_0)) +
geom_hex() +
geom_abline(intercept = 0)
課題
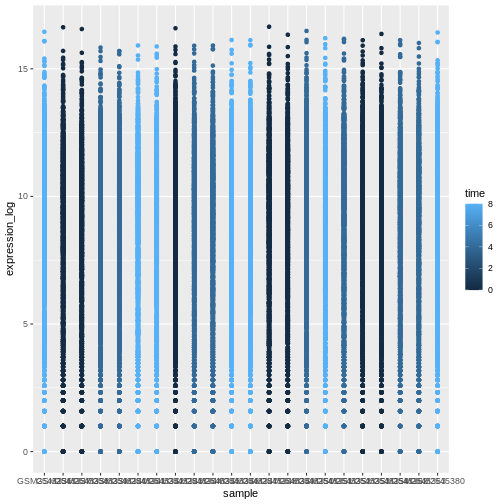
これまでに学んだ知識を活用して、rna データセットから
sample 列を選択し、expression_log
値を散布図としてプロットしてください。時間軸は異なる色で表示されるように設定します。このデータ表現方法はこの種のデータを示す上で適切な方法でしょうか?
R
ggplot(data = rna, mapping = aes(y = expression_log, x = sample)) +
geom_point(aes(color = time))
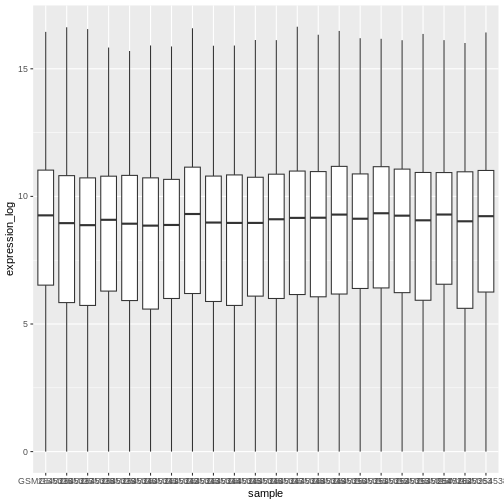
箱ひげ図
箱ひげ図を用いることで、各サンプル内における遺伝子発現量の分布を視覚的に表現することができます:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_boxplot()
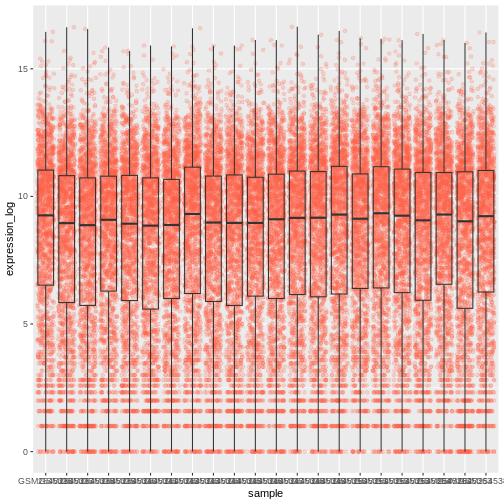
箱ひげ図に点プロットを追加することで、測定値の総数とその分布状況をより明確に把握することが可能になります:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_jitter(alpha = 0.2, color = "tomato") +
geom_boxplot(alpha = 0)
課題
箱ひげ図のレイヤーがジッタープロットのレイヤーの前面に表示されていることに注目してください。箱ひげ図を点の下に表示されるようにするには、コードをどのように変更すればよいでしょうか?
これら2つのジオメトリの順序を入れ替える必要があります:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_boxplot(alpha = 0) +
geom_jitter(alpha = 0.2, color = "tomato")
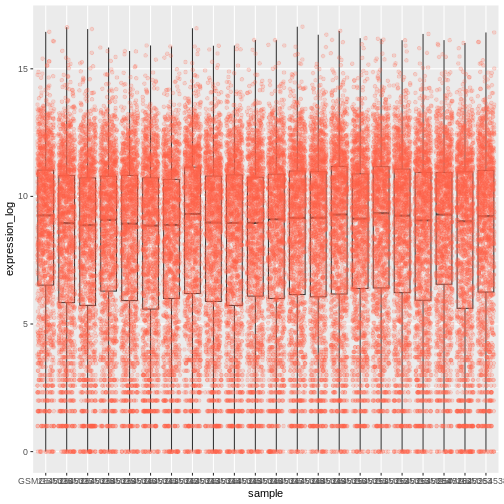
x軸上の値がまだ適切に読み取れないことにお気づきかもしれません。ラベルの表示方向を変更し、垂直方向と水平方向に調整して重なりが起こらないようにしましょう。90度の角度を使用するか、斜め向きのラベルに適した角度を見つけるために試行錯誤してみてください:
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_jitter(alpha = 0.2, color = "tomato") +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
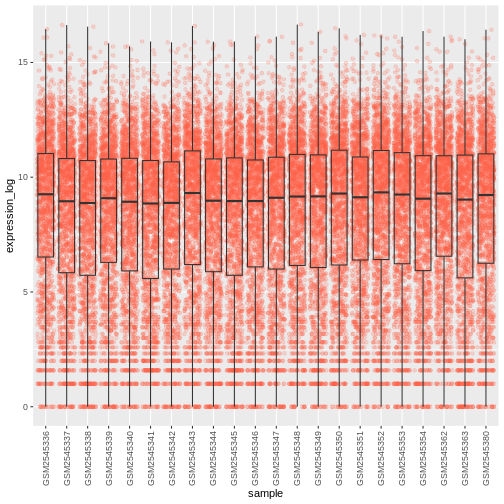
課題
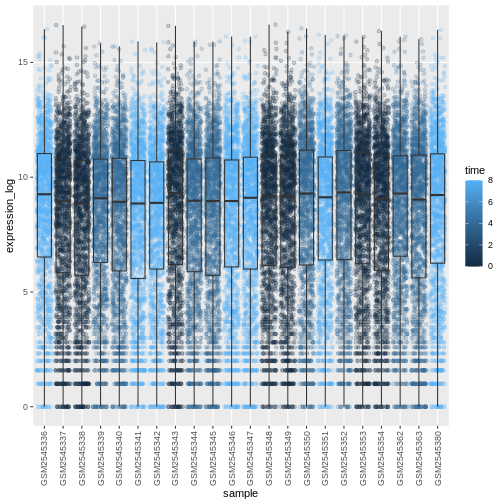
箱ひげ図のデータポイントに、感染期間(time)に応じて色を付けてください。
ヒント: time
変数のクラスを確認してください。ggplot のマッピング処理内で
time
のクラスを整数型から因子型に直接変更することを検討してください。この変更が
R でグラフを作成する方法にどのような影響を与えるのでしょうか?
R
# time as integer
ggplot(data = rna,
mapping = aes(y = expression_log,
x = sample)) +
geom_jitter(alpha = 0.2, aes(color = time)) +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
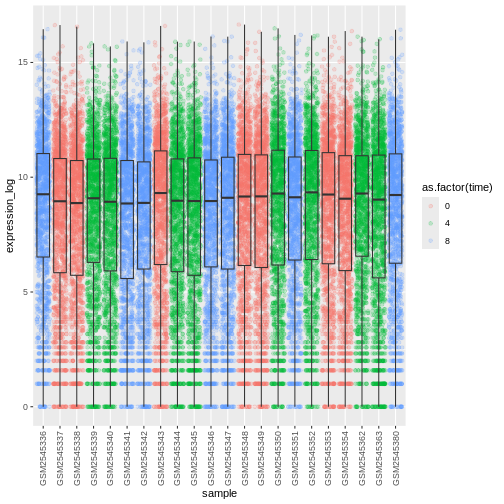
R
# time as factor
ggplot(data = rna,
mapping = aes(y = expression_log,
x = sample)) +
geom_jitter(alpha = 0.2, aes(color = as.factor(time))) +
geom_boxplot(alpha = 0) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
課題
箱ひげ図は分布の概要を把握するのに有用ですが、分布の「形状」そのものは表示しません。例えば、分布が二峰性の場合、箱ひげ図ではその特徴を捉えることができません。箱ひげ図の代替手段として「バイオリンプロット」があり、これはデータ点の密度分布の形状を視覚的に表現します。
- 箱ひげ図をバイオリンプロットに置き換えてください。
geom_violin()関数を使用します。fill引数を用いて、時間軸に沿ってバイオリンプロットを塗り分けてください。
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_violin(aes(fill = as.factor(time))) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
課題
- バイオリンプロットを修正し、
性別(sex)ごとにバイオリン部分を塗りつぶすようにしてください。
R
ggplot(data = rna,
mapping = aes(y = expression_log, x = sample)) +
geom_violin(aes(fill = sex)) +
theme(axis.text.x = element_text(angle = 90, hjust = 0.5, vjust = 0.5))
折れ線グラフ
感染期間ごとの平均発現量を、時間8時点と時間0時点を比較した際の対数倍率変化が最大となる10遺伝子について計算します。まず対象となる遺伝子を選択し、rnaデータセットからこれら10遺伝子のみを含むサブセットsub_rnaを作成します。その後、データをグループ化し、各グループ内の遺伝子発現量の平均値を算出します:
R
rna_fc <- rna_fc |> arrange(desc(time_8_vs_0))
genes_selected <- rna_fc$gene[1:10]
sub_rna <- rna |>
filter(gene %in% genes_selected)
mean_exp_by_time <- sub_rna |>
group_by(gene,time) |>
summarize(mean_exp = mean(expression_log))
OUTPUT
`summarise()` has grouped output by 'gene'. You can override using the
`.groups` argument.R
mean_exp_by_time
OUTPUT
# A tibble: 30 × 3
# Groups: gene [10]
gene time mean_exp
<chr> <int> <dbl>
1 Acr 0 5.07
2 Acr 4 5.54
3 Acr 8 7.31
4 Aipl1 0 3.70
5 Aipl1 4 3.89
6 Aipl1 8 6.56
7 Bst1 0 3.20
8 Bst1 4 3.77
9 Bst1 8 5.22
10 Chil3 0 4.00
# ℹ 20 more rows感染期間をx軸、平均発現量をy軸とした折れ線グラフを作成できます:
R
ggplot(data = mean_exp_by_time, mapping = aes(x = time, y = mean_exp)) +
geom_line()
残念ながらこの手法ではうまくいきません。すべての遺伝子のデータを一括してプロットしているためです。各遺伝子ごとに別々の線を描画するには、美的関数をgroup = geneに変更する必要があります:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp, group = gene)) +
geom_line()
色分けを追加すれば(colorパラメータを使用すると自動的にデータがグループ化されます)、プロット上で各遺伝子を区別できるようになります:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp, color = gene)) +
geom_line()
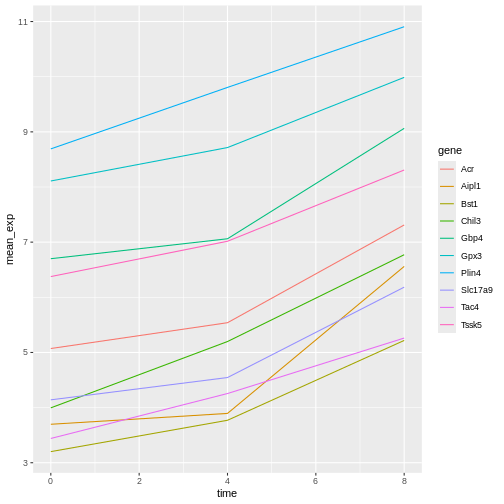
面分割表示
ggplot2には面分割と呼ばれる特殊な機能があり、データセットに含まれる因子に基づいて1つのプロットを複数のサブプロットに分割できます。これらのサブプロットは同じプロパティ(軸の範囲、目盛りなど)を継承するため、直接比較が容易になります。各遺伝子について時間経過に沿った折れ線グラフを作成する場合に活用しましょう:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp)) + geom_line() +
facet_wrap(~ gene)
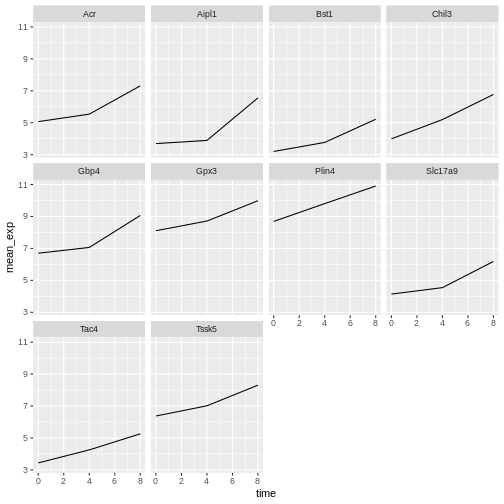
ここではすべてのサブプロットにおいて、x軸とy軸が同じスケールになっています。このデフォルト設定を変更するには、scalesパラメータを調整してy軸のスケールを自由に設定可能にできます:
R
ggplot(data = mean_exp_by_time,
mapping = aes(x = time, y = mean_exp)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y")
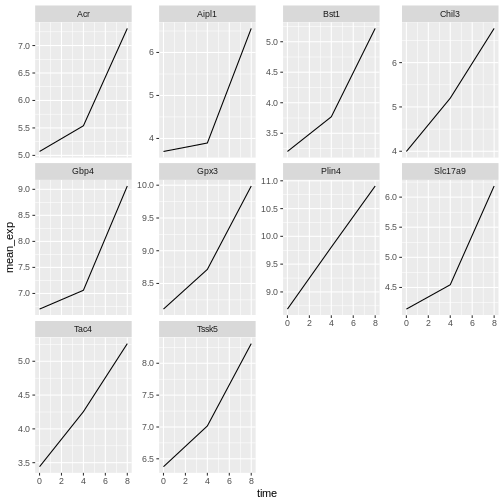
次に、各プロット内の線をマウスの性別でさらに分割したいとします。そのためには、gene、time、sexでグループ化したデータフレーム内で平均発現量を計算する必要があります:
R
mean_exp_by_time_sex <- sub_rna |>
group_by(gene, time, sex) |>
summarize(mean_exp = mean(expression_log))
OUTPUT
`summarise()` has grouped output by 'gene', 'time'. You can override using the
`.groups` argument.R
mean_exp_by_time_sex
OUTPUT
# A tibble: 60 × 4
# Groups: gene, time [30]
gene time sex mean_exp
<chr> <int> <chr> <dbl>
1 Acr 0 Female 5.13
2 Acr 0 Male 5.00
3 Acr 4 Female 5.93
4 Acr 4 Male 5.15
5 Acr 8 Female 7.27
6 Acr 8 Male 7.36
7 Aipl1 0 Female 3.67
8 Aipl1 0 Male 3.73
9 Aipl1 4 Female 4.07
10 Aipl1 4 Male 3.72
# ℹ 50 more rowsこれで、colorパラメータを使用して単一プロット内で性別ごとにさらに分割した面分割グラフを作成できます:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y")
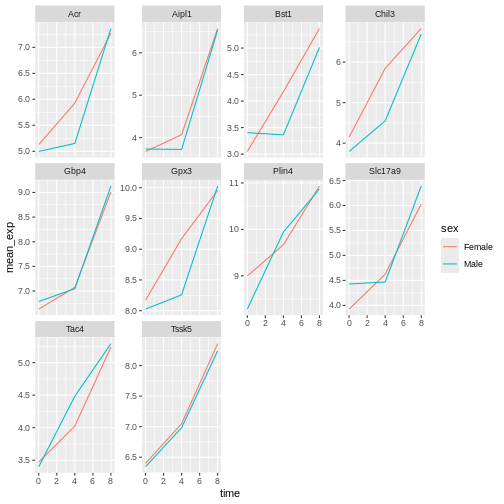
通常、白背景のプロットは印刷時に視認性が高くなります。theme_bw()関数を使用して背景を白に設定できます。さらに、グリッドラインも削除可能です:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
課題
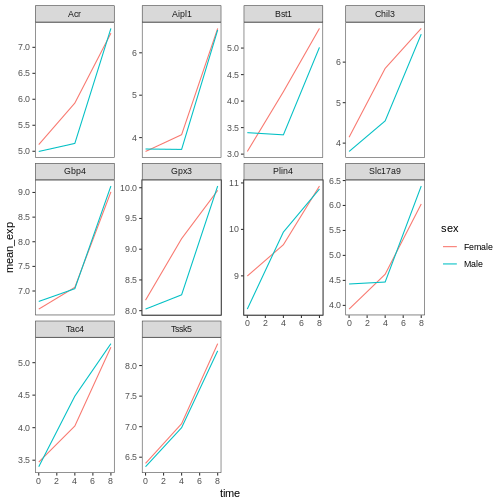
これまでに学んだ知識を活用して、感染経過に伴う各染色体の平均発現量の変化を視覚的に表現するプロットを作成してください。
R
mean_exp_by_chromosome <- rna |>
group_by(chromosome_name, time) |>
summarize(mean_exp = mean(expression_log))
OUTPUT
`summarise()` has grouped output by 'chromosome_name'. You can override using
the `.groups` argument.R
ggplot(data = mean_exp_by_chromosome, mapping = aes(x = time,
y = mean_exp)) +
geom_line() +
facet_wrap(~ chromosome_name, scales = "free_y")
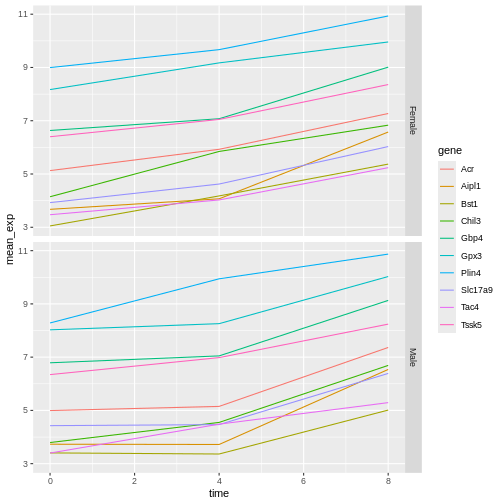
facet_wrapジオメトリはプロットを任意の数の次元に分割し、1ページにきれいに収まるようにします。一方、facet_gridジオメトリでは、数式表記(rows ~ columns;
単一の行または列を示すプレースホルダーとして.を使用可)を用いて、プロットの配置方法を明示的に指定できます。
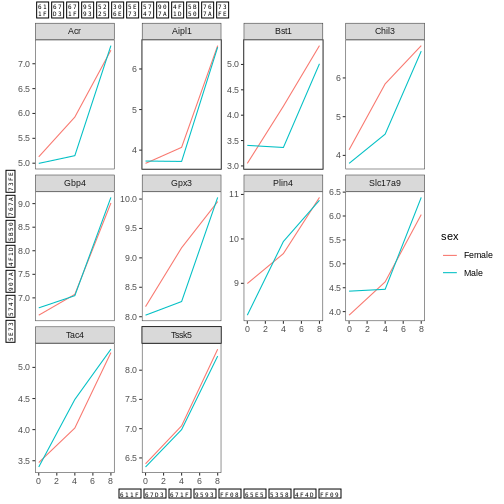
以前のプロットを修正して、性別ごとの遺伝子発現の平均値が時間経過とともにどのように変化したかを比較してみましょう:
R
# 1列レイアウト、行単位で分割
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = gene)) +
geom_line() +
facet_grid(sex ~ .)
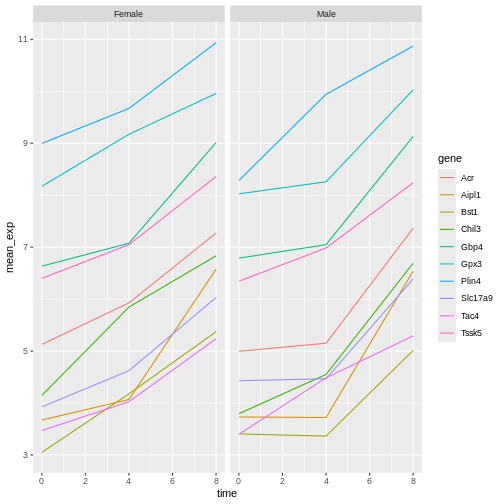
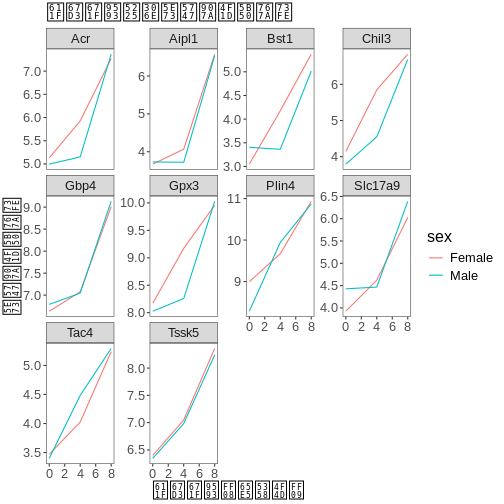
R
# 1行レイアウト、列単位で分割
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = gene)) +
geom_line() +
facet_grid(. ~ sex)
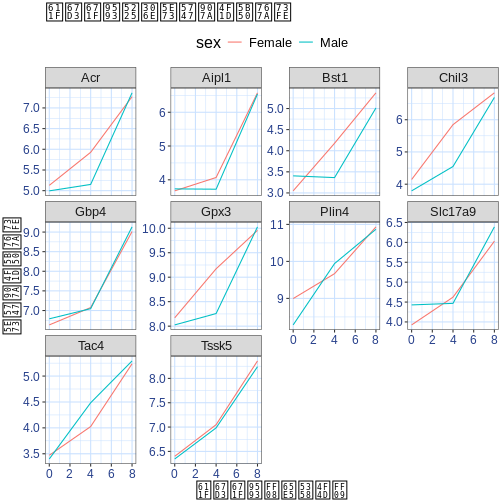
ggplot2テーマ
プロットの背景色を白に変更するtheme_bw()に加え、ggplot2には視覚化の外観を素早く変更できる他のテーマも用意されています。利用可能なテーマの完全なリストはhttps://ggplot2.tidyverse.org/reference/ggtheme.htmlで確認できます。theme_minimal()とtheme_light()は人気の高いテーマで、theme_void()は独自のテーマを作成する際の出発点として便利です。
ggthemesパッケージでは、Excel
2003テーマを含む多様なオプションが提供されています。ggplot2拡張機能
ウェブサイトでは、ggplot2の機能を拡張するパッケージの一覧が掲載されており、追加のテーマも含まれています。
カスタマイズ
時間と遺伝子ごとの平均発現量を性別で分割したプロットに戻りましょう。色分けは性別で行います。
ggplot2チートシートを参照し、プロットを改善する方法を考えてみてください。
次に、「時間」と「mean_exp」という単純な軸名をより情報量の多い表現に変更し、図にタイトルを追加しましょう:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "感染期間別の遺伝子発現平均値",
x = "感染期間(日数)",
y = "平均遺伝子発現値")
軸名はより情報量が増えましたが、可読性をさらに向上させるためにフォントサイズを大きくすることができます:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "感染期間別の遺伝子発現平均値",
x = "感染期間(日数)",
y = "平均遺伝子発現値") +
theme(text = element_text(size = 16))
なお、プロットのフォントを変更することも可能です。Windows環境では、extrafont
パッケージをインストールする必要がある場合があります。
さらに、x軸・y軸のテキスト色、グリッドの色などをカスタマイズできます。例えば、legend.positionを"top"に設定することで凡例を上部に移動させることも可能です。
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
labs(title = "感染期間別の遺伝子発現平均値",
x = "感染期間(日数)",
y = "平均遺伝子発現値") +
theme(text = element_text(size = 16),
axis.text.x = element_text(colour = "royalblue4", size = 12),
axis.text.y = element_text(colour = "royalblue4", size = 12),
panel.grid = element_line(colour="lightsteelblue1"),
legend.position = "top")
作成したカスタマイズテーマがデフォルトテーマよりも優れていると判断した場合、 このテーマをオブジェクトとして保存しておけば、今後作成する他のプロットに簡単に適用できます。以下に、以前に作成したヒストグラムを例に説明します。
R
blue_theme <- theme(axis.text.x = element_text(colour = "royalblue4",
size = 12),
axis.text.y = element_text(colour = "royalblue4",
size = 12),
text = element_text(size = 16),
panel.grid = element_line(colour="lightsteelblue1"))
ggplot(rna, aes(x = expression_log)) +
geom_histogram(bins = 20) +
blue_theme()
ERROR
Error in blue_theme(): could not find function "blue_theme"課題
ここまでの情報を踏まえ、この演習で生成されたプロットのいずれかを改善するか、
あるいはあなた自身の美しいグラフを作成するのにさらに5分間取り組んでください。
インスピレーションを得るために、RStudioのggplot2チートシート
を参照してください。以下にいくつかのアイデアをご紹介します:
- 線の太さを変更する方法を試してみてください。
- 凡例の名称を変更する方法はありますか?ラベルの変更はどうでしょう?
(ヒント:
scale_で始まるggplot関数を探してみてください) - 異なるカラーパレットを使用するか、線の色を手動で指定する方法を試してみましょう (詳細はhttps://www.cookbook-r.com/Graphs/Colors_(ggplot2)/をご覧ください)。
例えば、以下のプロットに基づいて:
R
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
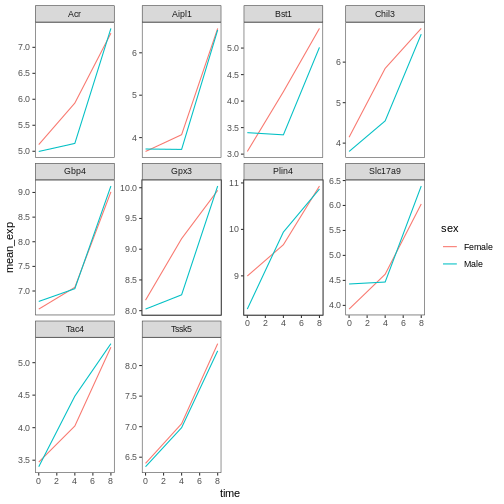
このプロットは以下の方法でカスタマイズ可能です:
R
# 線の太さを変更
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line(size=1.5) +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank())
WARNING
Warning: Using `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
ℹ Please use `linewidth` instead.
This warning is displayed once per session.
Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
generated.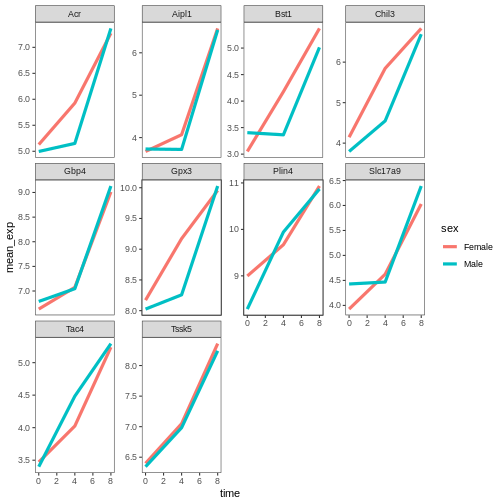
R
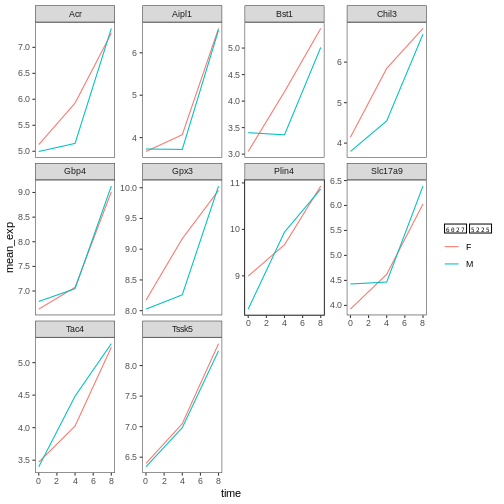
# 凡例名とラベルの名称を変更
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_discrete(name = "性別", labels = c("F", "M"))
R
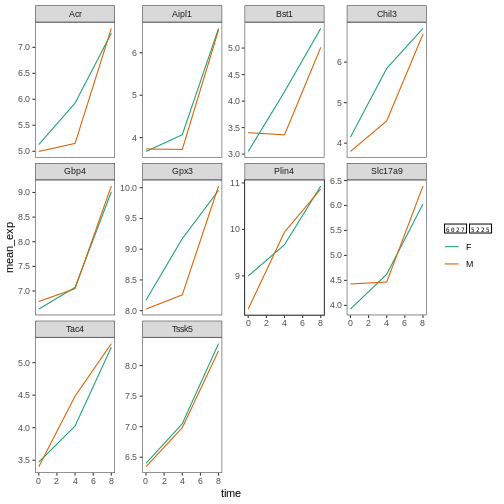
# 異なるカラーパレットを使用
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_brewer(name = "性別", labels = c("F", "M"), palette = "Dark2")
R
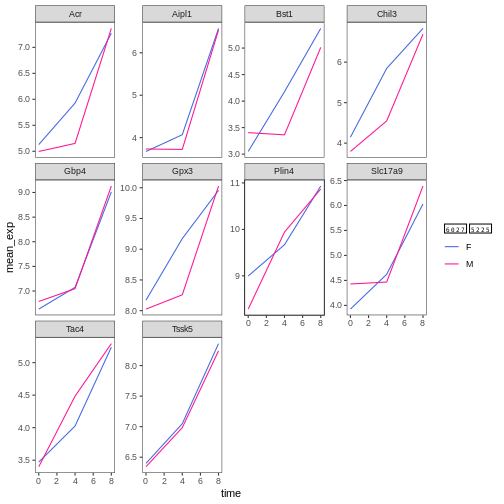
# 色を手動で指定
ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
theme_bw() +
theme(panel.grid = element_blank()) +
scale_color_manual(name = "性別", labels = c("F", "M"),
values = c("royalblue", "deeppink"))
複数プロットの配置方法
ファセット機能は1つのプロットを複数のサブプロットに分割する強力なツールですが、時には異なる変数やデータフレームに基づく独立した複数のプロットを1つの図にまとめたい場合もあります。
まず、横に並べて表示したい2つのプロットを作成しましょう:
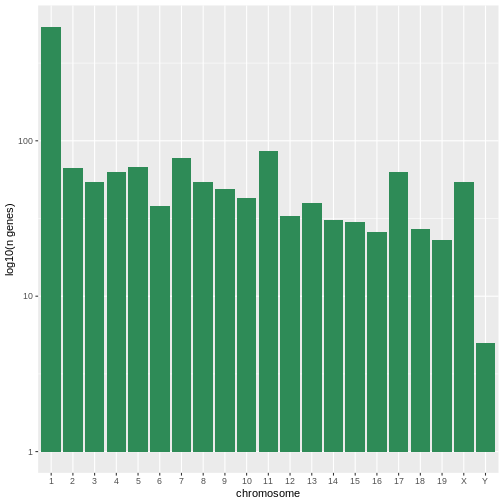
最初のグラフは各染色体ごとのユニークな遺伝子数をカウントしたものです。まずchromosome_nameのレベル順序を並べ替え、各染色体ごとのユニークな遺伝子をフィルタリングします。また、読みやすさを向上させるため、y軸のスケールを対数10スケールに変更します。
R
rna$chromosome_name <- factor(rna$chromosome_name,
levels = c(1:19,"X","Y"))
count_gene_chromosome <- rna |>
select(chromosome_name, gene) |>
distinct() |> ggplot() +
geom_bar(aes(x = chromosome_name), fill = "seagreen",
position = "dodge", stat = "count") +
labs(y = "log10(遺伝子数)", x = "染色体") +
scale_y_log10()
count_gene_chromosome
次に、legend.positionを"none"に設定することで、凡例を完全に非表示にします。
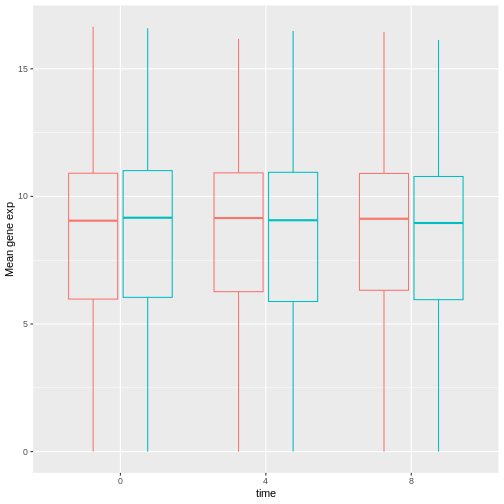
R
exp_boxplot_sex <- ggplot(rna, aes(y=expression_log, x = as.factor(time),
color=sex)) +
geom_boxplot(alpha = 0) +
labs(y = "平均遺伝子発現値",
x = "時間") + theme(legend.position = "none")
exp_boxplot_sex
patchworkパッケージは、+演算子を使用して図を整然と配置する洗練された方法を提供します。具体的には、|演算子で図を左右に並べ、/演算子で上下に重ねることが可能です。
R
install.packages("patchwork")
R
library("patchwork")
count_gene_chromosome + exp_boxplot_sex
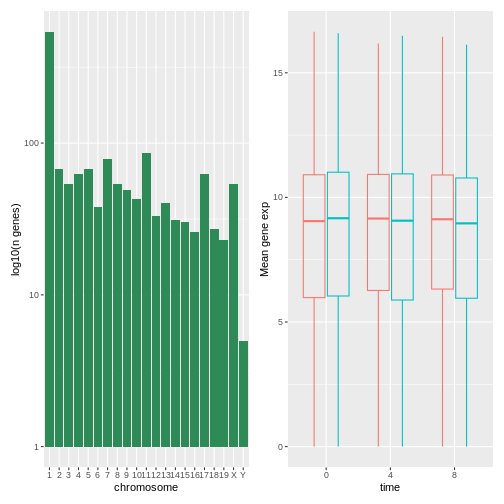
R
## または count_gene_chromosome | exp_boxplot_sex と記述することも可能
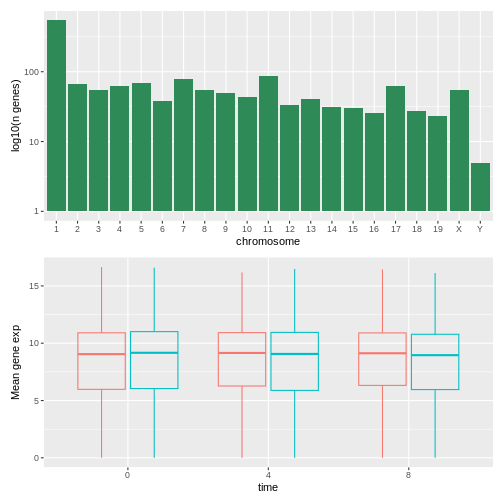
R
count_gene_chromosome / exp_boxplot_sex
plot_layout関数を使えば、さらに詳細なレイアウト制御が可能で、より複雑な配置も実現できます:
R
count_gene_chromosome + exp_boxplot_sex + plot_layout(ncol = 1)
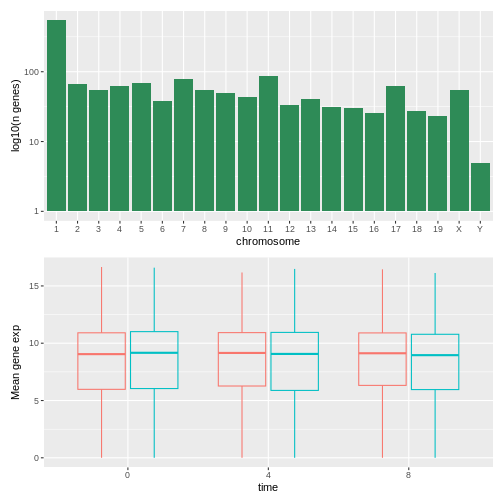
R
count_gene_chromosome +
(count_gene_chromosome + exp_boxplot_sex) +
exp_boxplot_sex +
plot_layout(ncol = 1)
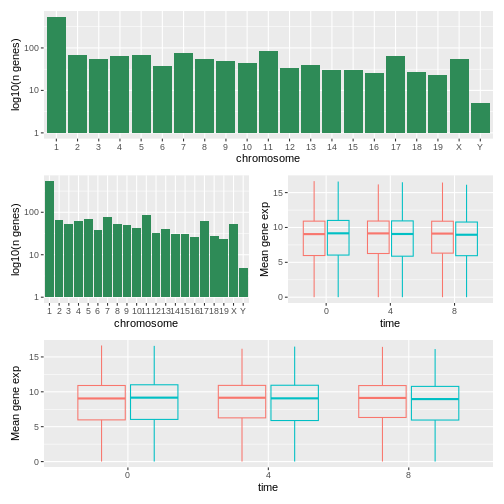
最後のプロット構成は、|と/の配置演算子を使っても作成できます:
R
count_gene_chromosome /
(count_gene_chromosome | exp_boxplot_sex) /
exp_boxplot_sex
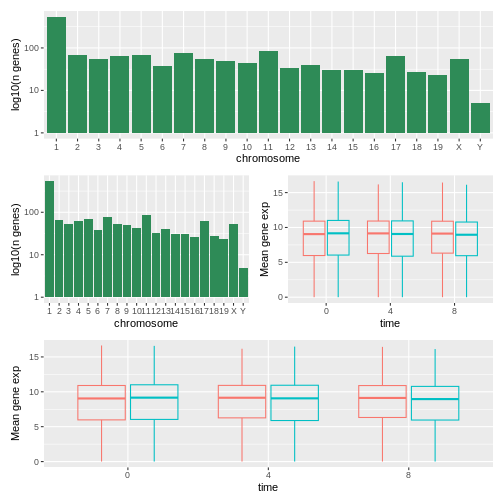
patchworkについてさらに詳しく知りたい場合は、公式ウェブサイトやこの解説動画をご覧ください。
別の選択肢として、gridExtraパッケージを使用する方法もあります。このパッケージではgrid.arrange()関数を使って個別のggplotプロットを1つの図に統合できます:
R
install.packages("gridExtra")
R
library("gridExtra")
grid.arrange(count_gene_chromosome, exp_boxplot_sex, ncol = 2)
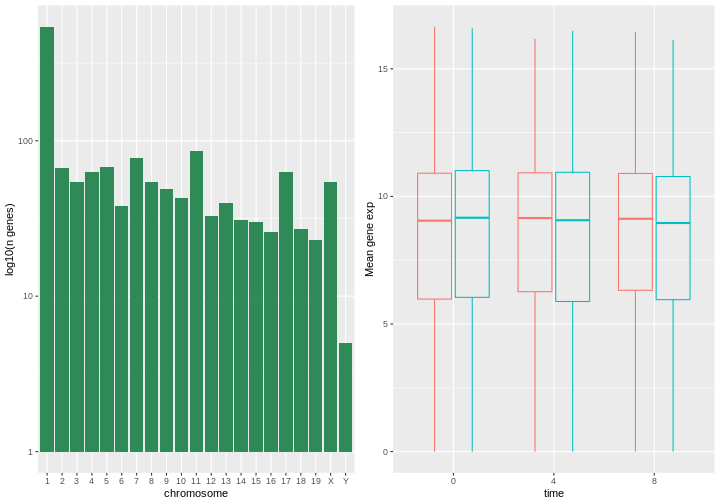
ncolとnrow引数を使った基本的な配置に加え、より複雑なレイアウト構成を実現するツールも用意されています。
プロットのエクスポート方法
プロットの作成が完了したら、お好みの形式でファイルに保存できます。RStudioの「プロット」ペインにある「エクスポート」タブを使用すると、低解像度でプロットを保存できますが、多くの学術誌では受理されず、ポスター用の拡大表示にも適さない場合があります。
代わりに、ggsave()関数を使用することをお勧めします。この関数では、適切な引数(width、height、dpi)を調整することで、プロットの寸法と解像度を簡単に変更できます。
作業ディレクトリにfig_output/フォルダが作成されていることを確認してください。
R
my_plot <- ggplot(data = mean_exp_by_time_sex,
mapping = aes(x = time, y = mean_exp, color = sex)) +
geom_line() +
facet_wrap(~ gene, scales = "free_y") +
labs(title = "感染期間別の平均遺伝子発現量",
x = "感染期間(日数)",
y = "平均遺伝子発現量") +
guides(color=guide_legend(title="性別")) +
theme_bw() +
theme(axis.text.x = element_text(colour = "royalblue4", size = 12),
axis.text.y = element_text(colour = "royalblue4", size = 12),
text = element_text(size = 16),
panel.grid = element_line(colour="lightsteelblue1"),
legend.position = "top")
ggsave("fig_output/mean_exp_by_time_sex.png", my_plot, width = 15,
height = 10)
# grid.arrange()で作成したプロットも同様に保存可能です
combo_plot <- grid.arrange(count_gene_chromosome, exp_boxplot_sex,
ncol = 2, widths = c(4, 6))
ggsave("fig_output/combo_plot_chromosome_sex.png", combo_plot,
width = 10, dpi = 300)
注意:widthとheightのパラメータは、保存されるプロットのフォントサイズも決定します。
可視化のためのその他のパッケージ
ggplot2は非常に強力なパッケージであり、私たちの「クリーンなデータ」と「クリーンなツール」のワークフローに非常によく適合します。Rには他に無視できない可視化パッケージも存在します。
基本グラフィックス
Rに標準で付属しているグラフィックスシステム、いわゆる「基本Rグラフィックス」はシンプルで高速です。これは「画家モデル」または「キャンバスモデル」に基づいており、異なる出力が直接重ね合わされます(図@ref(fig:paintermodel)参照)。これはggplot2(および後述するlattice)とは根本的な違いがあり、ggplot2は画面やファイルにレンダリングされる専用オブジェクトを返すため、さらに更新することも可能です。
R
par(mfrow = c(1, 3))
plot(1:20, main = "最初のレイヤー(plot(1:20)で作成)")
plot(1:20, main = "h = 10で追加した水平赤色線")
abline(h = 10, col = "red")
plot(1:20, main = "rect(5, 5, 15, 15)で追加した長方形")
abline(h = 10, col = "red")
rect(5, 5, 15, 15, lwd = 3)
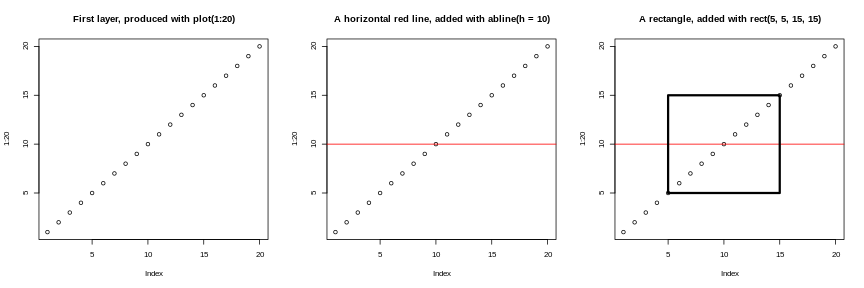
もう一つの重要な違いは、基本グラフィックスのプロット関数が入力データの種類に基づいて「適切な」処理を試みる点です。つまり、入力データのクラスに応じて動作を適応させます。これはggplot2とは大きく異なり、ggplot2はデータフレームのみを入力として受け付け、プロットを段階的に構築していく必要があります。
R
par(mfrow = c(2, 2))
boxplot(rnorm(100),
main = "rnorm(100)のボックスプロット")
boxplot(matrix(rnorm(100), ncol = 10),
main = "matrix(rnorm(100), ncol = 10)のボックスプロット")
hist(rnorm(100))
hist(matrix(rnorm(100), ncol = 10))
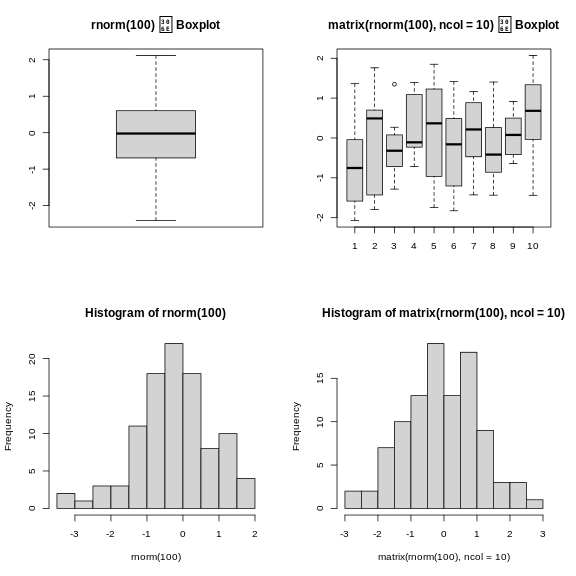
基本グラフィックスのデフォルト設定は、シンプルで標準的な図を素早く作成する場合に非常に効率的です。plot、hist、boxplotなどの単一の関数と1行のコードで迅速に生成できます。ただし、デフォルト設定が常に最も見栄えが良いとは限らず、特に複雑な図(例えばファセット表示を作成する場合など)では、調整に時間がかかり煩雑になることがあります。